Introduction
Generation of a maximally diverse gene library is particularly important when employing non-targeted mutagenesis strategies. The method most often used to generate variants with random mutations is error-prone PCR (epPCR). Here we present the basic protocols and tips for using epPCR to generate gene variants that exhibit a relatively balanced spectrum of mutations and for capturing as much diversity as possible through effective cloning of those variants.
The principles of epPCR
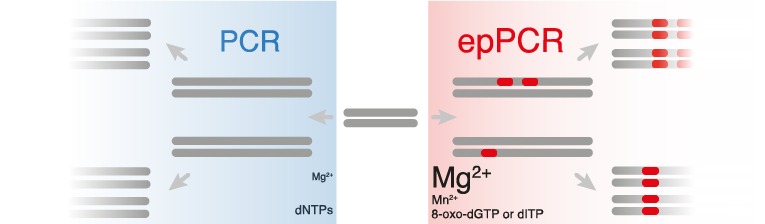
Error-prone PCR protocols are modifications of standard PCR methods, designed to alter and enhance the natural error rate of the polymerase. There are many ways to control the mutation rate of epPCR.
- EpPCR reactions typically contain higher concentrations of MgCl2 (7 mM) compared to basic PCR reactions (1.5 mM), in order to stabilize non-complementary pairs.(Ling et al., 1991)
- MnCl2 can also be added to increase the error-rate.(Lin-Goerke, Robbins & Burczak, 1997)
- Other ways of modifying mutation rate include varying the ratios of nucleotides in the reaction or including a nucleotide analog such as 8-oxo-dGTP or dITP. Mutation frequencies from 0.11 to 2% (1 to 20 nucleotides per 1 kb) have been achieved simply by varying the nucleotide ratio and the amount of MnCl2 in the PCR reaction.(Fromant, Blanquet & Plateau, 1995)
- The number of genes that contain a mutation can also be modified by changing the number of effective doublings by increasing/decreasing the number of cycles or by changing the initial template concentration.
An expression system and high-throughput assay should be developed before a library of enzyme variants are generated. epPCR provides access to an almost unlimited number of variants, but generally have a lower hit rate and are therefore better suited to high throughput screening or selection strategies. To take full advantage of the power of error-prone PCR, the assay must be accurate enough to detect small improvements and sensitive enough to detect the low levels of activity typically encountered in the beginning rounds of an evolution experiment.
The procedures of epPCR
-
epPCR Reaction Preparation*
For each PCR sample, add to tube:
· 10 μL 10X normal error-prone PCR buffer ()
· 2 μL 50X dNTP mix
· Additional dNTPs [optional; increase mutation rate]
· 10 μL 55 mM MgCl2 [optional; increase mutation rate]
· 10 μL 55 mM MnCl2 [optional; increase mutation rate]
· 30 pmol each primer [design your primers before]
· 2 fmol template DNA (~10 ng of an 8-kb plasmid) [Prepare your template before]
· 1 μL Taq polymerase (5U)
· H2O to a final volume of 100 μL.
There are also some commercial available epPCR Kits: (Agilent)[https://www.agilent.com/en/product/mutagenesis-cloning/mutagenesis-kits/random-mutagenesis-kits/genemorph-ii-233115], (TakaraBio)[https://www.takarabio.com/products/cloning/mutagenesis-kits/random-mutagenesis-kit] -
Run Error-Prone PCR Program
· 30 s at 94°C
· 30 s at annealing temperature for primers
· 1 min at 72°C (for a ~1 kb gene)
· 35-50 cycles (more cycles can increase mutations)
· 5 min at 72°C final extension
· 4°C (to protect samples overnight if necessary) -
Construct the library
Using Gibson Assembly or Goldengate Assembly for library construction.
3.1 Gibson Assembly(Gibson et al., 2009) -
Gibson Mix Buffer(2x) 5μl
- Vector backbone 25 fmol
- Each insert 50 fmol
- H20 To make 10 μl
50℃, 1 hour.
-
Goldengate Assembly
Type IIs RE 0.50μl
T4 ligase 0.50μl
Vector backbone 25 fmol
Each insert 50 fmol
Goldengate buffer 1 μl
H20 To make 10 μl -
Step 1, RE digestion
37℃(BsaI) or 42℃(BsmaI) 2 min - Step 2, Ligation
16 °C 5 min -
Step 3, RE digestion
60 °C 10 min
Step 1-3 repeat 25 cycles
Step 4 Heat inactivation
80 °C 10 min
Hold
4 °C
∞ -
Transformation
Prepare a high-efficiency cell library like E. coli or yeast for transformation. (key step). See more information: Transformation of genetic library. -
Selection
Using a designed selection method for selection. More information is coming soon.
Very comprehensive protocol for students:
https://experiments.springernature.com/articles/10.1007/978-1-4939-1053-3_1
References
Fromant, M., Blanquet, S. & Plateau, P. (1995) Direct Random Mutagenesis of Gene-Sized DNA Fragments Using Polymerase Chain Reaction. Analytical Biochemistry. [Online] 224 (1), 347–353. Available from: doi:10.1006/abio.1995.1050.
Gibson, D.G., Young, L., Chuang, R.-Y., Venter, C.J., et al. (2009) Enzymatic assembly of DNA molecules up to several hundred kilobases. Nature Methods. [Online] 6 (5), nmeth.1318. Available from: doi:10.1038/nmeth.1318.
Ling, L.L., Keohavong, P., Dias, C. & Thilly, W.G. (1991) Optimization of the polymerase chain reaction with regard to fidelity: modified T7, Taq, and vent DNA polymerases. Genome Research. [Online] 1 (1), 63–69. Available from: doi:10.1101/gr.1.1.63.
Lin-Goerke, J.L., Robbins, D.J. & Burczak, J.D. (1997) PCR-Based Random Mutagenesis Using Manganese and Reduced dNTP Concentration. BioTechniques. [Online] 23 (3), 409–412. Available from: doi:10.2144/97233bm12.
- Technical Notes
- Open Access
- Published: 30 January 2012
Microbial Cell Factories
volume 11, Article number: 14 (2012)
Cite this article
-
14k Accesses
-
13 Citations
-
1 Altmetric
-
Metrics details
Abstract
Background
Error-prone PCR (epPCR) libraries are one of the tools used in directed evolution. The Gateway® technology allows constructing epPCR libraries virtually devoid of any background (i.e., of insert-free plasmid), but requires two steps: the BP and the LR reactions and the associated E. coli cell transformations and plasmid purifications.
Results
We describe a method for making epPCR libraries in Gateway® plasmids using an LR reaction without intermediate BP reaction. We also describe a BP-free and LR-free sub-cloning method for in-frame transferring the coding sequence of selected clones from the plasmid used to screen the library to another one devoid of tag used for screening (such as the green fluorescent protein). We report preliminary results of a directed evolution program using this method.
Conclusions
The one-step method enables producing epPCR libraries of as high complexity and quality as does the regular, two-step, protocol for half the amount of work. In addition, it contributes to preserve the original complexity of the epPCR product.
Background
Gateway® is an appealing technology because its cloning efficiency is close to 100% [1]. This feature is particularly welcome when dealing with numerous target genes, for instance in Structural Genomics. Unfortunately, high throughput gene expression following gene cloning in Structural Genomics programs has also revealed that many recombinant proteins are insoluble in E. coli thereby precluding their crystallization and their study by X-ray crystallography. Among the different techniques used to overcome this insolubility problem, one is directed evolution. The use of directed evolution for improving recombinant protein solubility can be summarised as follows. A random library of mutants generated by error-prone PCR (epPCR) and/or DNA shuffling [2] is screened for variant proteins more soluble than the wild-type (wt) protein. To that end, the mutated DNA sequences may be expressed as fusion proteins with a C-terminal «solubility reporter» such as the green fluorescent protein (GFP) [3]. To assess the solubility gain provided by the mutations, the mutated coding sequences are then sub-cloned from the solubility reporter expression plasmid to a GFP-free expression plasmid and the solubility of the tag-free variant is compared to that of the tag-free wt protein expressed under the same conditions.
Although the Gateway® technology is less used in directed evolution than in Structural Genomics programs, it has been nevertheless successfully applied in a directed evolution study that made use of both epPCR and DNA shuffling [4]. The evolved Tobacco Etch Virus (TEV) protease exhibited significantly higher solubility than the wt TEV protease. Incidentally, this study also revealed a few weak points that seemed to be specifically associated with the use of the Gateway® technology rather than with the screening process or the protein to evolve. In particular, (i) the number of expression clones was found to be relatively small, as also reported in another study [5]; (ii) the generation of epPCR and DNA shuffling libraries was labor intensive because of the need for BP and LR reactions to be carried out, and of the corollary transformations and intermediate plasmid medium preparations [6]; (iii) the subcloning of the coding sequence of selected mutants from the reporter expression plasmid to a non-reporter expression plasmid was also time-consuming because of the same requirements.
While the first of the drawbacks listed above can be easily addressed by transforming expression cells by electroporation, addressing the other two requires devising a novel cloning and sub-cloning strategy. With the specific purpose of overcoming these limitations while maintaining the obvious advantages of the Gateway® technology, we devised a method that allows eliminating the BP step from the generation of the library and both the BP and LR steps from the sub-cloning process. We applied this method to generate a diversity library of the intrinsically disordered C-terminal domain of the measles virus nucleoprotein (NTAIL) [7, 8] as a first step towards the dissection of the molecular mechanisms underlying its interaction with the C-terminal X domain (XD, aa 459-507) of the viral phosphoprotein [9–17]. A split-GFP reassembly assay [18–20] was used to screen the library and to identify clones with novel binding properties.
Results
1) Generation of an epPCR library
The conventional procedure for generating epPCR libraries using the Gateway® technology comprises two recombination reactions (BP and LR) [4]. We first addressed the question as to whether each recombination reaction and associated E. coli cell transformation decreased the complexity of a given library. A typical Gateway® recombination reaction can be described as the transfer of an insert from a donor to a non-recombined acceptor to yield a recombined acceptor. Therefore, the library complexity loss can be evaluated by comparing the number of colonies provided by: ( i ) a theoretical experiment made of a 100% efficient LR reaction (i.e. a reaction where all the non-recombined acceptor (i.e., Gateway® plasmid before LR reaction) molecules are used to yield recombined acceptors (i.e., Gateway® plasmids after LR reaction)) followed by a 100% efficient cell transformation (i.e. a transformation where all recombined acceptor molecules are uptaken by cells and where each cell uptakes one recombined acceptor molecule); (ii) an actual cell transformation by a recombined acceptor; (iii) an actual cell transformation by an actual LR reaction using the same donor construct (i.e., the other substrate of the LR reaction) and the same non-recombined acceptor as in the previous two instances. The results of this comparison are reported in Table 1. Since 25 fmoles of acceptor correspond to 1.55 × 1010 molecules, if the LR reaction and cell transformation were each 100% efficient, then one should obtain 1.55 × 1010 colonies per 25 fmoles of input acceptor. However, transforming E. coli cells by electroporation with 25 fmoles of recombined acceptor provided a mean value of 18.7 × 107 colonies. Assuming that each cell uptakes only one plasmid molecule, this means that E. coli cells electroporation was responsible for a ~80-fold drop of the theoretical (maximal) number of clones in this experiment. In addition, when E. coli cells were electroporated with an LR reaction mixture using the same amount of the same acceptor, the average number of colonies was 2.7 × 107 (Table 1). This means that the recombination reaction was by itself responsible for an additional ~7 fold efficiency drop. Incidentally, this latter comparison provided a direct measure of the efficiency of the LR reaction, which could be determined to be approximately 14% under the conditions used in this experiment. In conclusion, each step made of a recombination reaction followed by transformation of E. coli by electroporation reduced the number of clones with respect to what could be expected from a 100% efficient LR reaction combined with a 100% efficient cell transformation. Thus, skipping one recombination reaction and the associated E. coli transformation in the Gateway® cloning process would be expected to better preserve the original library complexity than the classical two step approach (BP followed by LR), particularly when heat-shock is used instead of electroporation to transform E. coli cells (Table 1). Incidentally, this would also reduce the risk of biasing the pENTR library (i.e., the BP reaction product, see stage 1 in the left panel of Figure 1) by unbalanced clone growing during the culture in the presence of kanamycin. On the basis of this observation, we devised a new «single recombination» method for constructing epPCR libraries.
Full size table
Full size table
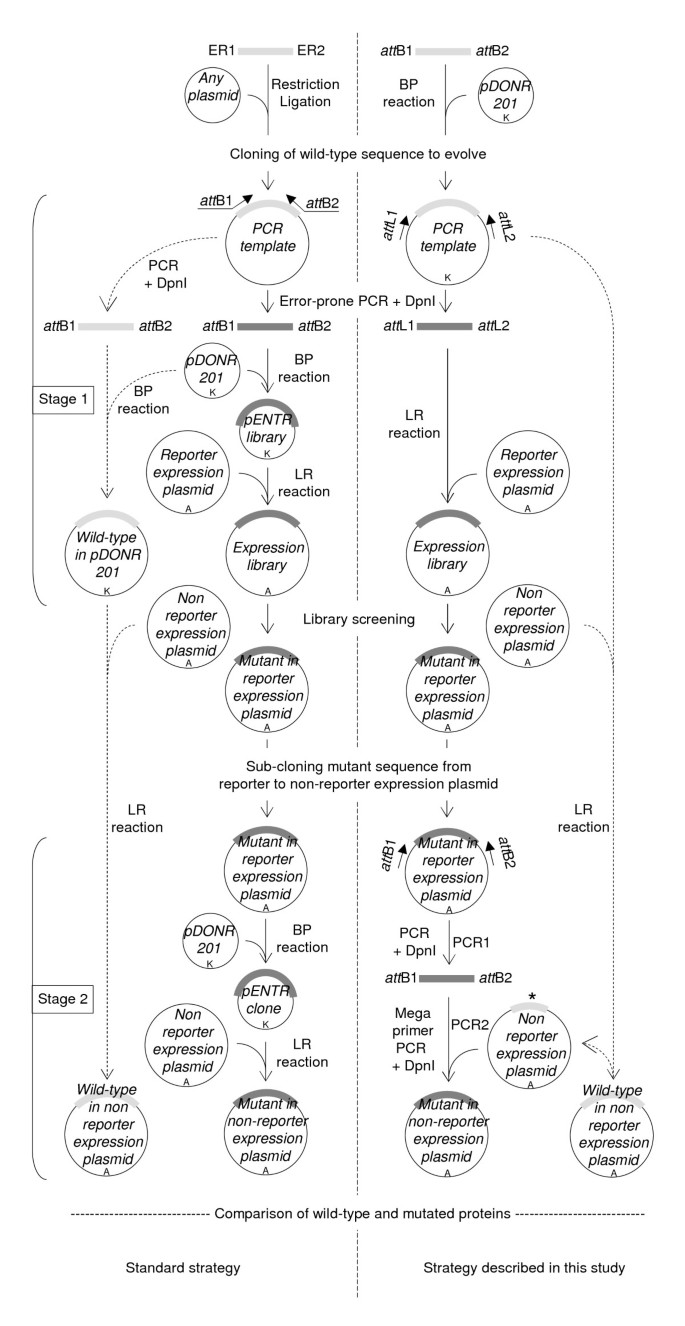
Overview of the method. The left flowchart is the standard strategy and the right flowchart is the strategy described in this study. Brackets on the left indicate the two stages of the strategy: the epPCR library construction (stage1) and the sub-cloning of mutant inserts from reporter to non-reporter expression plasmid (stage 2). ER1 and ER2 denote restriction sites used to clone the sequence to evolve and create the PCR template of the standard strategy. Inner arrows with continuous lines are the core of the method. Outer arrows with dashed lines indicate the pathways used to transfer the wt sequence to the non-reporter expression plasmid, and (in the right flowchart only) to create the internally deleted wt NTAIL in pDEST17O/I. Mutated and wt sequences are represented by thick dark and light grey lines, respectively. The internally deleted wt sequence is denoted by an asterisk. Antibiotic resistance markers are indicated: A, ampicillin resistance; K, kanamycin resistance.
Full size image
The strategy we have devised is described in Figure 1 (right flowchart) and compared to the regular strategy (left flowchart and reference [4]). It includes not only the generation of the library (Figure 1, stage 1), but also the sub-cloning of selected mutant sequences from the expression plasmid used for screening the random mutant library (referred to as «reporter expression plasmid» throughout the text) to a different expression plasmid (referred to as «non-reporter expression plasmid») (Figure 1, stage 2) allowing the variant proteins of interest to be expressed without the screening tag (such as GFP). In the regular strategy [4], generating and then cloning an epPCR library into a Gateway® reporter expression plasmid requires three steps (Figure 1, stage 1, left flowchart). In the first step, the epPCR product (dark grey in Figure 1, stage 1) is generated using two primers hybridizing to the 5′ and 3′ ends of the coding sequence desired to be subjected to epPCR and flanked at their respective 5′ end by Gateway®att B1 and att B2 recombination sites (25-mer), and a PCR template consisting of the wt target sequence (light-grey in Figure 1, stage 1). In the second step, the epPCR product is inserted in a shuttle plasmid (pDONR201) by mean of a BP reaction. After E. coli transformation, an aliquot is spread on kanamycin plates to assess the library complexity, and the remaining is grown in liquid culture (100 ml). A plasmid medium preparation is then performed to recover the cloned library (pENTR library) from the liquid culture. In the third step, an aliquot of the plasmid medium preparation is used in an LR reaction to transfer the inserts from the entry clones of the pENTR library to a reporter expression plasmid. The LR reaction is performed as described above for the BP reaction except that ampicillin is used instead of kanamycin. In this procedure, obtaining a high-complexity library relies on the efficiency of two critical steps, namely the BP and LR reactions. In the strategy we developed, the epPCR library is generated using the att L1 (25-mer) and att L2 (24-mer) primers and the wt coding sequence already cloned in the pDONR201 plasmid as PCR template. Primers att L1 and att L2 respectively hybridize to the «Forward- and Reverse-priming sites» located upstream and downstream the att L1 (100 bp) and att L2 (100 bp) Gateway® recombination sites (Additional file 1: Figure S1). As a result, the epPCR product is flanked by full-length att L1 and att L2 recombination sites and hence can be directly used in the LR reaction. This procedure enables to eliminate three steps of the regular approach: (i) the BP reaction that transfers the epPCR product to pDONR to create the pENTR library; (ii) the transformation of E. coli with the pENTR library; (iii) the purification (plasmid medium preparation) of the latter. Beyond the advantage related to the reduction of the number of steps, this procedure also offers the advantage of preserving the library complexity as shown above.
2) Sub-cloning of selected mutated sequences
After mutants of interest have been selected, their coding sequence must generally be sub-cloned from the reporter expression plasmid used to screen the epPCR library to another non-reporter expression plasmid (Figure 1, stage 2). In the regular strategy [4], this procedure essentially follows the same process as that used to create the random library (compare stage 1 and stage 2 of Figure 1, left flowchart). In the first step, the mutated coding sequence is transferred from the reporter expression plasmid to pDONR201 by mean of a BP reaction. E. coli cells are transformed with the BP reaction mixture, and transformed cells are selected on kanamycin plates. Given the efficiency of Gateway® transfer [1], few colonies need to be analyzed. The plasmid of each colony is purified by mini-preparation, and then checked for the presence of the proper insert generally by PCR using the att L1 and att L2 primers. The plasmid of one positive pENTR clone is then used as substrate in an LR reaction to transfer the coding sequence from pDONR to the non-reporter expression plasmid. The regular strategy works well when few mutants have to be processed, but we realized that it was impractical in directed evolution projects where many mutated coding sequences have to be sub-cloned in parallel from reporter to non-reporter expression plasmid(s). Therefore, we devised the alternative strategy depicted in the right flowchart of Figure 1, stage 2 and in more details in Figure 2. This strategy could be described as a hybrid method between MEGAWHOP [21] and RF cloning [22]. MEGAWHOP was devised to clone a random library of linear DNA. In practice, a library of mutated linear DNA is used as a complementary pair of megaprimers in a PCR experiment. The PCR template is the non mutated DNA sequence borne by the expression plasmid to be used for screening the library. Since the hybridization mismatch between the wt sequence borne by the expression plasmid and each mutated sequence to sub-clone (i.e., the mutation rate) is low compared to the homology, the megaprimers hybridize very efficiently to the wt sequence. During the PCR elongation steps, the whole plasmid is copied. After amplification, the PCR template is degraded by DpnI treatment. The final product is the random library of DNA borne by the expression plasmid. RF cloning also aims at inserting a linear double strand DNA into a plasmid, but uses a different approach. In contrast to MEGAWHOP, not the whole length of the linear DNA to clone is used for hybridization, but only 24 base pairs at both ends which are designed to be complementary to the cloning site of the plasmid. When this linear DNA is used in a PCR experiment with the cloning plasmid as template, each 24 base flanking extension hybridizes to its complementary sequence on the plasmid, resulting in a linear amplification of the plasmid during the PCR elongation step. After amplification the PCR template is degraded by DpnI treatment. The final product is the linear DNA borne by the plasmid. Our sub-cloning technique relies on the use of megaprimers, as in MEGAWHOP, and on the annealing of only the ends of the megaprimers, as in RF cloning. The method consists of two steps. In the first step, the mutated coding sequence is PCR amplified using the att B1 and att B2 primers and the reporter expression plasmid bearing the mutated coding sequence as template (PCR1, Figures 1 and 2). After DpnI treatment to remove methylated (i.e. parental) DNA, the PCR product flanked by full-length att B1 and att B2 Gateway® recombination sites is used as a pair of complementary megaprimers in a second PCR step that uses an internally deleted form of the wt coding sequence borne by the non-reporter expression plasmid as template (PCR2, Figures 1 and 2). The reason for using an internally deleted sequence as PCR2 template is explained in the last paragraph of the results section. The result of this second PCR is the full-length mutated coding sequence in the non-reporter expression plasmid.

Schematic of the sub-cloning of a mutated sequence from reporter to non-reporter expression plasmid. From top to bottom, the DNA fragments amplified by PCR between the T7prom and att B2 primers are 1,046 bp, 333 bp, and 560 bp in length, respectively. On the right is illustrated how inverse PCR was used to internally delete 227 bp from the wt NTAIL sequence in pDEST17O/I. Plasmids are not at scale.
Full size image
3) Application of the method to the NTAIL-XD interaction project
We have applied the above-described strategy to few directed evolution projects (B. Coutard, F. Vincent, unpublished data), including the one reported herein. In view of gaining insights into the NTAIL-XD interaction, we generated a mutant library of NTAIL and used a split GFP re-assembly assay to screen variants with altered interaction abilities [18–20]. In this method, the sequence coding for one of the two interacting partners under study is inserted in the prokaryotic expression vector pET11a-link-NGFP so as to lead to its expression as a fusion protein with the N-terminal half of GFP (NGFP). The coding sequence of the second interacting partner is inserted in another prokaryotic expression vector (pMRBAD-link-CGFP) thus leading to its expression as a fusion protein with the C-terminal half of GFP (CGFP). Both fusion proteins are then co-expressed in E. coli. If the two proteins of interest interact with each other in the bacterium, their interaction allows the two GFP halves to re-associate and reconstitute the functional (i.e., fluorescent) GFP. Thus, fluorescent bacteria denote an interaction between the two partners, with the fluorescence intensity being proportional to the affinity of this interaction. In practice, a library of NTAIL random mutants was inserted in a modified pET11a-link-NGFP vector (see below), and mutants were screened for altered interacting abilities with wt XD expressed from pMRBAD-XD-CGFP.
Since neither pET11a-link-NGFP nor pMRBAD-link-CGFP are Gateway® plasmids, as a first step in this project we modified pET11a-link-NGFP to make it a Gateway® reporter expression plasmid. This conversion is generally performed by inserting a blunt-end synthetic cassette that is flanked by att R1 and att R2 recombination sites, and bears two constitutive genes: a chloramphenicol resistance (Cmr) gene, and a counter-selectable (ccd B) gene. The cassette can be purchased from Invitrogen in three reading frames, and is intended to be inserted in a blunt (or blunted) restriction site of the plasmid to modify. However, such a restriction site may not exist at the desired position, and blunt end cloning requires screening not only for cassette-containing plasmids but also for the correct orientation of the cassette with regard to the promoter. Since pET11a-link-NGFP cloning site contains two restriction sites (XhoI and BamHI), an alternative to the classical approach was to PCR amplify the cassette using whatever Gateway® destination plasmid as template and primers hybridizing to the 5′ and 3′ ends of the cassette and flanked by XhoI and BamHI restriction sites, respectively. Unfortunately, because of the high homology between the 5′ and 3′ ends of the Gateway® cassette (Figure 3A), each primer was found to be able to hybridize to both ends of the cassette (Figure 3B). To circumvent this problem, we devised the strategy depicted in Figure 3C. To prevent hybridization of the 5′ primer with the 3’primer site and vice versa, the 5′ and 3′ halves of the cassette were PCR amplified separately (PCR tube 1 and PCR tube 2, Figure 3C) using the Gateway® destination plasmid pTH31 [4] as template. This template contained a single BamHI restriction site in the middle of the cassette which had to be mutated before cloning. We took advantage of its central location to develop a pair of internal overlapping primers (primers 2 and 3) encoding a mutated BamHI site to mutate the internal BamHI restriction site during the amplification of the two cassette halves. Internal PCR primers 2 and 3 were 100% complementary to each other, so that the PCR products from tube 1 and tube 2 overlapped. After DpnI treatment to remove the template, and purification of the PCR products to remove PCR primers, a third PCR was run that only used an equimolar amount of the two PCR products, i.e., without adding external primers nor template (PCR tube 3, Figure 3C). This «elongation PCR» reconstituted the full-length cassette which was then ligated into pET11a-link-NGFP after BamHI and XhoI digestion. This Gateway® reporter expression plasmid was called pNGG (p lasmid N—G FP G ateway®) and is available upon request. Incidentally, this «two halves» strategy is generally applicable whenever direct directional cloning is hampered by a high homology of the two PCR primers.
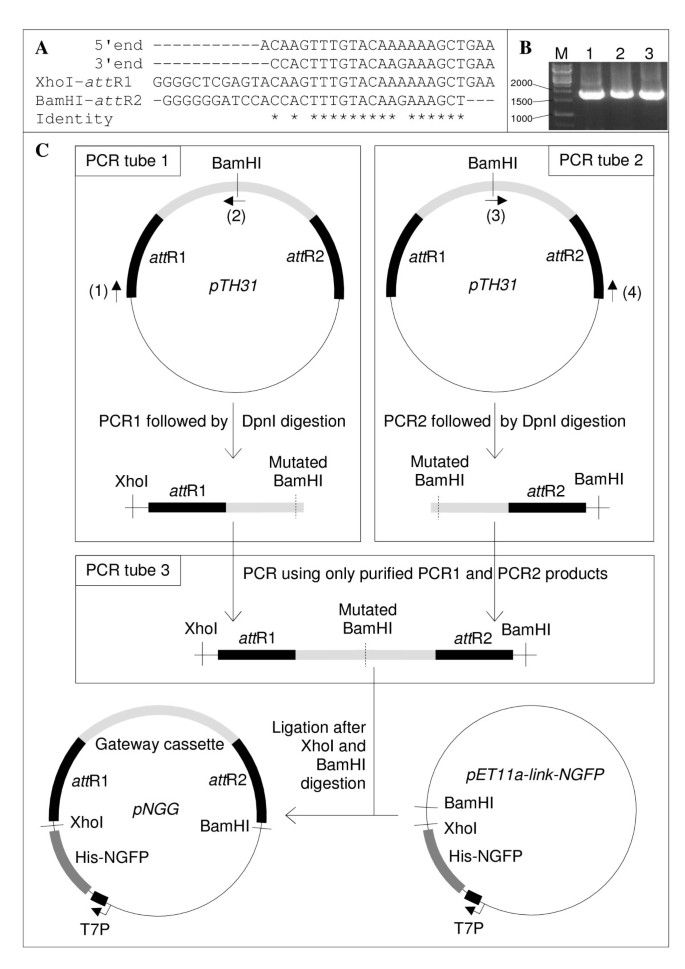
Construction of pNGG. (A) Alignment of (from top to bottom): the 5′ and 3′ ends of pTH31 Gateway® cassette; XhoI-att R1 PCR primer; BamHI-att R2 PCR primer. Sequence identity is denoted by asterisks below the alignment. (B) The Gateway® cassette was PCR amplified using pTH31 as template, and either primer XhoI-att R1 alone (lane 1), primer BamHI-att R2 alone (lane 2), or primers XhoI-att R1 and BamHI-att R2 (lane 3). Markers size is indicated on the left in base pairs. (C) «Two-halves» making of pNGG. The plasmids are not at scale. Light grey, Gateway cassette. Black, att R recombination sites. Primer (1), XhoI-att R1. Primer (2), BamHI-mut-R. Primer (3), BamHI-mut-F. Primer (4), BamHI-att R2 (Table 3).
Full size image
Full size table
In a first step, the efficiency of LR reactions using different donors and acceptors was evaluated under different experimental conditions. We first assessed the efficiency of an LR reaction that made use of a non-mutated linear molecular species bearing att L recombination sites at the extremities (wt NTAIL-PCR, which could be described as a «ghost pENTR clone» since it is devoid of most of the pENTR backbone) and a circular Gateway® reporter expression plasmid (non-linearized pNGG), and compared it to that of LR reactions that were carried out with different combinations of linear and non-linear substrates. The results are reported in Table 4. The combination used in the strategy we devised, i.e. wt NTAIL-PCR and non-linearized pNGG (Table 4, line 3), provided at least as good results as the other combinations, in particular when compared to the reference wt NTAIL-pDONR/non-linearized pNGG combination (Table 4, line 1). Thus, a linear DNA flanked by full-length att L recombination sites proved to be an efficient substrate for the LR reaction. We also checked whether the use of non-equimolar ratios of either LR reaction substrates, as suggested by Invitrogen, was optimal. The results, reported in Table 3, indicated that 100 fmoles of the donor construct and 25 fmoles of the acceptor construct provided at least as good results as an equimolar amount (100 fmoles of each), suggesting that 25 fmoles of non-linearized pNGG did not limit the LR reaction efficiency under these experimental conditions. The strategy we devised relies on the use of a pair of generic att L primers to generate an epPCR product flanked by the full-length att L1 and att L2 recombination sites (Stage 1 in the right flowchart of Figure 1). Since each att L recombination site is 100 bp long (Additional file 1: Figure S1), a risk potentially existed that mutations had been introduced by Mutazyme II in these sites during epPCR elongation steps. This would lead to an epPCR product partly unsuitable for the LR reaction, thereby ultimately resulting in a library complexity drop. As one can see in the first two lines of Table 5, 100 and even 10 fmoles of NTAIL-epPCR product provided at least as many clones as did 100 fmoles of NTAIL-PCR product generated by a proof-reading Taq polymerase (Table 4 third line, and Table 3 first line). This suggests that if mutations occurred in the att L recombination sites during the epPCR, they did not interfere with the downstream LR reaction. This result is not completely surprising since it has been shown that deleting as much as 50% of att L recombinant site did not significantly reduce the percentage of recombinant clones [23]. Finally, we investigated the reproducibility of our approach. As shown in Table 5 (last three lines), the results obtained with NTAIL/pNGG were not unique to this system, as comparable results were obtained when coding sequences with a composition and length (LadS, PA3059) different from that of NTAIL were used in LR reactions with another reporter expression plasmid (pTH31). This suggested that using a linear DNA generated by epPCR flanked by att L sites, and a non-linearized plasmid in the LR reaction was a generally applicable method.
Full size table
Full size table
In the next step, we evaluated the ability of our method for generating epPCR libraries (Stage 1 in the right flowchart of Figure 1) to provide mutants of interest. To that end, an epPCR library of NTAIL in pNGG (Table 5, second line) was screened for clones displaying higher or lower fluorescence with respect to that of wt NTAIL when co-expressed with XD as described in Methods. More than 300 clones complying with these criteria were manually selected and characterized in terms of their fluorescence and of their sequence (Gruet et al., unpublished results). The latter revealed an average mutation rate of 1% base pairs. Attempts to increase this rate by performing additional epPCRs failed for the reasons explained in Additional file 2: Figure S2. Figure 4A shows the fluorescence of a representative sample of the selected clones. Antiparallel leucine zippers (Z) (pET11a-Z-NGFP and pMRBAD-Z-CGFP) were used as positive control of interaction [18] because of their high affinity, expression, and solubility. An NTAIL coding sequence with an in-frame stop codon located just downstream att B1 was used as negative control (Stop-NTAIL, S). This construct expresses only the NGFP moiety which is unable to interact with XD-CGFP. The reference fluorescence value was provided by wt NTAIL (N). A representative set of 4 mutants (1-4) with a fluorescence similar to or lower than that of wt NTAIL is reported. The relationship between the mutations borne by these mutants (Additional file 3: Text S1) and their specific fluorescence is beyond the scope of this study and will be discussed elsewhere (Gruet et al., unpublished results). Notably, clone 4 featured a stop codon (R489 (CGA) > (TGA) Stop) that resulted in a truncated form of NTAIL. Interestingly, this deletion perfectly mimics an already published variant (NTAILΔ2,3) that had been shown to display a considerably lower (two orders of magnitude) affinity towards XD [10]. Since we could not rule out a priori that a decreased fluorescence could be due to decreased protein expression and not to decreased NTAIL-NGFP/XD-CGFP interaction, we analyzed the total and soluble fractions of the different NTAIL-NGFP clones (S, N, 1-4). Taking advantage of the presence of a 6His tag appended at the N-terminus of NGFP [18], the His-tagged proteins expressed by clones Z, S, N, and 1-4 were purified by immobilized metal (Ni2+) affinity chromatography (IMAC) from the total E coli lysate under denaturing conditions, or from the soluble fraction of this latter under non denaturing conditions. The eluants from IMAC were analyzed by SDS-PAGE (Figure 4B). Notably, Stop-NTAIL, wt NTAIL, and all four mutants exhibited comparable levels of total expression of the NGFP-NTAIL fusion protein (arrows 1 and 2 in Figure 4B) suggesting that fluorescence differences were due to different interaction abilities and not to different protein expression levels. Moreover, the expression level of the NGFP-NTAIL protein by the non-fluorescent mutant 4 was even slightly higher than that of the other mutants (arrow 2 in Figure 4B). Under non denaturing conditions, the interacting partner fused to CGFP and devoid of His-tag (arrows 5 and 6 in Figure 4B) was co-purified with the His-tagged protein. As expected, its amount paralleled the fluorescence intensities reported in Figure 4A. By contrast, when proteins were purified by IMAC under denaturing conditions, only the His-tagged moiety was recovered from total E coli lysate. Altogether, these results indicate that different NTAIL-XD interaction capabilities due to specific mutations in the NTAIL sequence and not different NTAIL expression levels accounted for the different fluorescence intensities reported in Figure 4A. Thus, our one step method for generating epPCR libraries (Stage 1 in the right flowchart of Figure 1) proves to be an effective mean to yield mutants of interest.
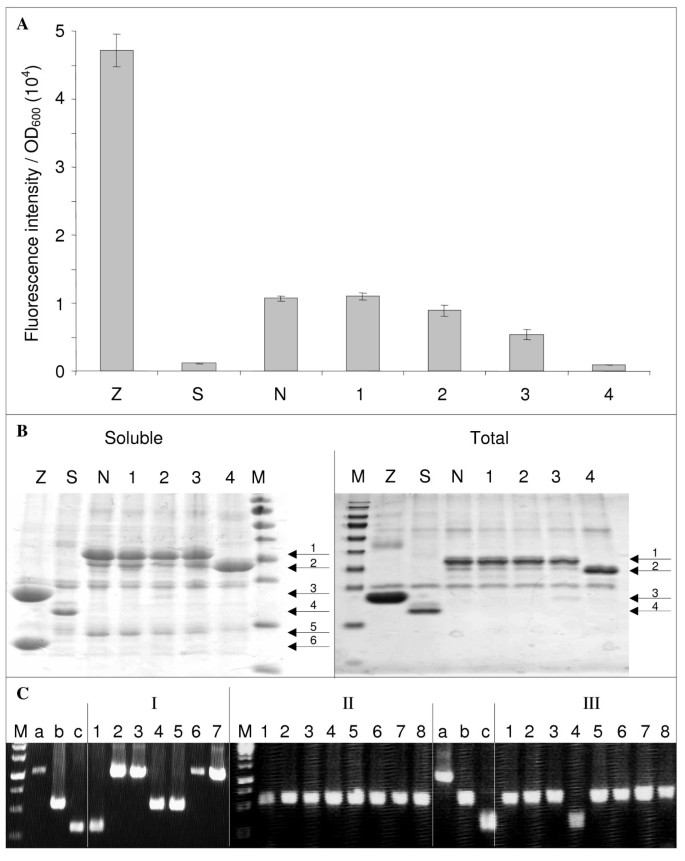
Representative results of library screening and sub-cloning experiments. (A) The fluorescence to OD600 ratio (mean value and standard deviation of a triplicate experiment) of the clones indicated on the × axis were determined as described in Methods. Z, Leucine zippers; S, Stop-NTAIL; N, wt NTAIL; 1-3, full length mutated NTAIL; 4, truncated NTAIL mutant. (B) His-tagged proteins expressed by clones Z, S, N, 1-4 (Figure 4A) were purified by affinity chromatography on IMAC as described in Methods, and were analyzed by SDS-PAGE using 15% polyacrylamide gels and Coomassie blue staining. Soluble, His-tagged proteins were purified under non denaturing conditions from the soluble fraction of the E. coli lysate. Total, His-tagged proteins were purified under denaturing conditions from total E. coli lysate. Soluble and total fractions were obtained from a duplicate culture. M, molecular size markers (from top to bottom: 170, 130, 100, 70, 55, 40, 35, 25, 15, 10 kDa). Arrows indicate the different purified proteins: 1, NGFP- wt NTAIL and NGFP-full-length NTAIL variants (34 kDa); 2, NGFP-truncated NTAIL variant 4 (29.4 kDa); 3, NGP-Z (22.8 kDa); 4, NGFP (i.e., Stop-NTAIL) (20.4 kDa); 5, XD-CGFP (15.5 kDa); 6, Z-CGFP (13.3 kDa). (C) PCR screening of mutated NTAIL sub-cloning experiment from pNGG to pDEST17O/I. PCR control a, 1,046 bp; b, 560 bp; c, 333 bp. PCR screening II and III were run in the same gel along with controls a, b, and c. M, molecular size markers (from top to bottom: 2000, 1500, 1000, 750, 500, 250 bp).
Full size image
In view of investigating interaction capabilities in the absence of the GFP moiety (to be published elsewhere), the mutated NTAIL coding sequences were sub-cloned from pNGG (reporter expression plasmid) to pDEST17O/I (non-reporter expression plasmid), a frame-compatible plasmid allowing proteins to be expressed with a simple N-terminal His-tag [24]. Sub-cloning was performed as described above (Figure 1, stage 2, right panel and Figure 2). The efficiency of this approach is highly dependent on the efficiency of DpnI treatment performed after each PCR. An inefficient DpnI treatment after PCR1 would generate a background made of the donor construct (mutant sequence in pNGG). An inefficient DpnI treatment after PCR2 would generate a background made of the acceptor construct (wt sequence in pDEST17O/I). Unfortunately, this signal-to-noise ratio could be appraised only by sequencing plasmids borne by several randomly chosen clones. To avoid sequencing PCR1 or PCR2 background, we introduced a screening step so as to be able to distinguish background plasmids from the desired construct. The easiest way to do that was to use a pair of screening primers that amplify fragments of different lengths from the three possible constructs (i.e., the correct construct, and PCR1 and PCR2 backgrounds). Although a primer pair made of T7prom and att B2 primers could distinguish pNGG from pDEST17O/I, it failed to differentiate a mutated NTAIL from the wt NTAIL sequence in pDEST17O/I, as both have the same length. A solution to this issue was to use as template in PCR2 an internally deleted wt NTAIL sequence (Figure 2) instead of the full-length sequence as envisaged by MEGAWHOP [21]. The internally deleted (227 bp deletion) NTAIL borne by pDEST17O/I (pDEST17O/I-idNTAIL, Table 2) was constructed by inverse PCR using the pDEST17O/I-NTAIL plasmid (Table 2) as template and primers p17O/ISal1F and p17O/ISal1R (Table 6). Following DpnI digestion, the PCR product was purified and then digested with SalI and self-ligated (Figure 2). Under these conditions, PCR screening E. coli colonies after transformation with DpnI-treated PCR2 product using T7prom and attB2 primers was expected to lead to amplification of either a 1,046 bp band (PCR1 background), or a 560 bp band (correct construct), or a 333 bp band (PCR2 background). Typical PCR screening results are reported in Figure 4C. First experiments (experiment I in Figure 4C) revealed a low signal (i.e., correct construct) to background ratio. In gel I, clone 1 was PCR2 background, while clones 2, 3, 6 and 7 were PCR1 background and only clones 4 and 5 contained the correct construct. The high background (5 clones out of 7) denoted inefficient DpnI treatments, particularly after PCR1 (4 clones out of 7). This issue was addressed in two different ways. Firstly, we reduced the background by improving DpnI treatment and by reducing the amount of the template used in PCR1 as the PCR template is responsible for the background. Secondly, as the signal is proportional to (and limited by) the amount of megaprimers used in PCR2 we increased the signal by increasing the amount of megaprimers produced by PCR1. In practice, to both increase the signal and decrease the background we combined several modifications of the reaction: more DpnI was used for longer incubation times, more of each primer, more PCR cycles, larger PCR volumes and less PCR template were used in experiments II and III (Additional file 4: Table S1) to generate the megaprimers (PCR1). In experiment II, the template used for PCR2 was full-length NTAIL in pDEST17O/I. The new experimental conditions (Additional file 4: Table S1) completely abolished PCR1 background (no band detectable at 1,046 bp in Figure 4C, experiment II). However, the use of full-length NTAIL in pDEST17O/I as template in PCR2 did not allow detecting PCR2 background. Therefore, an experiment was performed as in experiment II except that internally deleted NTAIL in pDEST17O/I was used as template in PCR2. The results (experiment III in Figure 4C) confirmed the absence of PCR1 background and pointed out a low level (similar to that of experiment I although PCR2 template had been increased, see Additional file 4: Table S1) of PCR2 background. PCR-positive clones 1, 2, 3 and 5 from experiment III were sequenced, and proved to bear the correct insert in the correct plasmid. In addition, sequencing ruled out the presence of any unwanted mutations that could have been introduced because of the increase (from 10 to 30) in the number of cycles in PCR1.
Full size table
Discussion
In this study, we described a new method for creating high complexity epPCR libraries based on a modified use of the Gateway® recombination cloning technology. Compared to the conventional Gateway® protocol ([4] and stage 1 in the left flowchart of Figure 1), our method is faster as it skips some steps such as the BP reaction and the associated E. coli transformation and plasmid purification. Note that although we only used epPCR for generating random mutant libraries, we believe that our strategy could be easily applied to the generation of libraries based on DNA shuffling [2]. Since the conventional sub-cloning of selected mutated sequences requires the same steps as those typically used to create the library ([4] and stage 2 in the left flowchart of Figure 1), we also devised a sub-cloning strategy that allows several mutant sequences to be sub-cloned in parallel from the reporter expression plasmid used to screen the library to another expression plasmid devoid of the screening tag (GFP in this case). We have applied and validated the method in three directed evolution projects, and provided here the first results obtained in the case of the NTAIL-XD system. In the other two projects, we constructed other epPCR libraries that made use of other target sequences to evolve, as well as of another reporter expression plasmid. In all cases, comparable library complexities were obtained (Table 5 and unpublished data).
As shown in Table 1, skipping the BP reaction used in the standard protocol to clone the epPCR product in pDONR (stage 1 in Figure 1) appears to preserve the library complexity. However, one may argue that the price paid for this preservation is that the wt sequence must first be cloned into pDONR, and so the benefit of our strategy (i.e. the reduction in the number of steps) would be abrogated by this first mandatory «pre-cloning» BP reaction. We would like to point out that this is not the case. This starting construct is instrumental not only in the epPCR step but also, directly or indirectly, in three other steps of our procedure (see dashed line in the right flowchart of Figure 1). Firstly, this pDONR construct bearing the wt sequence is required to generate by LR reaction the pDEST17O/I-wt NTAIL construct (i.e. the non-reporter expression plasmid bearing the wt sequence) that will be used as a control in comparative expression experiments with the selected mutated sequences (Figure 1). Secondly, pDEST17O/I-wt NTAIL allows the construction of the internally deleted template used in the sub-cloning (Figure 2). Thirdly, pDEST17O/I-wt NTAIL also provides a positive control in PCR screenings aimed at avoiding the sequencing of background clones (Figure 4C). By contrast, since the standard strategy (stage 1 in the left flowchart of Figure 1) used a non Gateway® plasmid as template for the epPCR, the same result will require much more work and steps (compare dashed lines in the left and right flowcharts of Figure 1). Thus, what could first appear as a constraint finally proves to be a saving of time when compared to the standard protocol.
RF cloning and MEGAWHOP could not be used in our sub-cloning protocol exactly as they were published, and needed to be adapted for the following reasons. In the published RF cloning technique [22], PCR1 was performed using genomic DNA as template, and so inefficient DpnI digestion would simply result in bacteria being transformed with genomic DNA. Since the latter did not provide any antibiotic resistance it could not generate any background, whereas the template was responsible for PCR1 background in our case (Figure 4C). In the case of MEGAWHOP [21], donor and acceptor plasmids were the same. Here again, inefficient DpnI digestion of PCR2 product would only increase the amount of non mutated sequences in the final epPCR library, a situation frequently encountered even at high mutagenesis rates (A. Gruet, unpublished observations), whereas it was responsible for PCR2 background in our case (Figure 4C).
It is noteworthy that the sub-cloning method that we developed was made possible because of the following two features. Firstly, donor (pNGG) and acceptor (pDEST17O/I) plasmids were both Gateway® plasmids and hence shared common 5′ and 3′ PCR priming sites. Moreover, these PCR priming sites did not require specific but generic att B1 and att B2 primers. Secondly, thanks to the specific founding features of Gateway®, pNGG and pDEST17O/I were 5′ and 3′ reading frame compatible, a feature that can be easily obtained for any expression plasmid suitably modified to bear the Gateway® cassette. For example, we have taken advantage of this feature to sub-clone mutated coding sequences from pTH31 to pTH24 [4] in another directed evolution project (B. Coutard, unpublished results).
Although it does simplify sub-cloning when more than five mutant coding sequences have to be processed in parallel, the sub-cloning strategy that we devised could be further optimized by using a PCR2 template that could be detected directly on plates. This would save the time devoted to pick up and grow randomly chosen clones and to purify and screen their plasmids so as to distinguish them from those from background colonies. Actually, we sought at using Rubredoxin [25] as such a possible phenotypic marker of PCR2 background. Unfortunately however, following transformation of E. coli with a pDEST17O/I plasmid bearing the sequence of a synthetic Thermotoga maritima Rubredoxin gene (NCBI reference sequence: NP_228468.1) that had been optimized for translation in E. coli, no red colonies were obtained (data not shown). Another even more stringent mean would be to use a ccd B constitutive gene in pDEST17O/I instead of an internally deleted NTAIL[26]. In that case, there would be no PCR2 background as clones bearing the ccd B gene are not viable.
Before concluding, we would like to point out that the method described here is proposed as a toolbox made of three independent parts: (i) the production of epPCR libraries, (ii) the «two halves» construction of a Gateway® plasmid, (iii) a sub-cloning strategy. The NTAIL-XD project provided an opportunity to describe a combined usage of the three parts, but these can be used separately (B. Coutard, unpublished results).
The Gateway® cloning technology has been a tremendous breakthrough since it was implemented in the early 2000s. Perhaps not by chance, its commercial availability happened to coincide with the beginning of the Structural Genomics era. This coincidence undoubtedly helped Structural Genomics programs to comply with their time schedule [1]. To our surprise, we have been unable to find numerous directed evolution projects that made use of the Gateway® technology. We hope that the modified protocol described in this study will contribute to a broader usage of this powerful technology in studies aimed at evolving proteins.
Conclusions
We have described a method to create high complexity epPRC libraries using only the LR reaction of the Gateway® cloning technology. Directly cloning an epPCR product in the plasmid used for screening the library allows eliminating not only the BP reaction but also the associated E. coli cell transformation and plasmid purification required by the usual Gateway® procedure. Thus, the same result can be obtained with half the amount of work.
Methods
Materials
Turbo broth (TB) was from AthenaES™. NucleoSpin® Extract II and NucleoSpin® Plasmid kit (Macherey-Nagel) were used for purifying PCR products and for plasmid mini-preparations, respectively. DNA ligations were performed using Quick Ligation Kit (Roche). GeneMorph® II Random Mutagenesis Kit (Stratagene) was used for epPCR experiments. Preparative and analytical PCRs were carried out using Pfx and Taq polymerase (Invitrogen), respectively. All primers were purchased from Operon. DNA sequencing was performed by GATC Biotech.
DNA constructs
pMRBAD-link-CGFP [19], encoding the C-terminal half (aa 158-238) of eGFP under the control of the PBAD promoter (inducible by L-arabinose) and allowing upstream in frame cloning of DNA fragments, was a kind gift of Lynne Regan, as were pET11a-link-NGFP, pET11a-Z-NGFP and pMRBAD-Z-CGFP. These latter two encode fusion proteins between leucine zippers and the two halves of the eGFP, while pET11a-link-NGFP encodes the N-terminal half (aa 1-157) of eGFP under the control of the T7 promoter (inducible by IPTG) and allows downstream in-frame cloning of DNA fragments [19]. pMRBAD-XD-CGFP was constructed as follows. The coding sequence of XD was PCR amplified using primers Nco1-MeV.XD and MeV.XD-AatII (Table 6) and a plasmid encoding the X domain (aa 459-507) of the measles virus phosphoprotein (strain Edmonston B) (pDEST14/XDHC) as template [9]. DpnI-treated purified PCR product and pMRBAD-link-CGFP were ligated after NcoI and AatII digestion. pDONR-NTAIL was obtained by BP reaction with a PCR NTAIL fragment amplified using primers NTAILF and NTAILR (Table 6), and a plasmid encoding the measles virus nucleoprotein (strain Edmonston B) (pET-21a/N) as template [27]. pDONR-Stop-NTAIL was obtained by BP reaction with a PCR NTAIL fragment amplified using primers StopNtail and NTAILR (Table 6) and pDONR-NTAIL as template. StopNtail has two Stop codons before the first NTAIL codon. pNGG-NTAIL and pNGG-Stop-NTAIL were obtained by LR reaction using pNGG as destination vector and pDONR-NTAIL or pDONR-Stop-NTAIL as donor constructs, respectively. pDEST17O/I-NTAIL was obtained by LR reaction using pDONR-NTAIL and pDEST17O/I [24]. The constructs were verified by sequencing and found to conform to expectations. The plasmids used in this study are summarized in Table 2.
Strains and electroporation
DB3.1 cells (Invitrogen) were used to propagate non recombined Gateway® plasmids, and TAM1 cells (Active Motif) to propagate non-Gateway® and recombined Gateway® plasmids. T7 cells (New England Biolabs) bearing the pLysS plasmid from Rosetta(DE3)pLysS cell (Novagen) (referred to as T7pRos) were used for protein expression and for epPCR library screening.
Electrocompetent T7pRos cells were prepared as follows. Frozen cells that had been previously transformed with pMRBAD-XD-CGFP were used to seed 1 L of LB containing 50 μg/ml kanamycin and 34 μg/ml chloramphenicol, and allowed to grow until OD600 = 0.5. The cells were recovered by centrifugation for 10 min at 5000 g, and resuspended in 400 ml of ice-cold water. Three additional washings were performed using 300 and then 200 ml of ice-cold water. The cells were then washed with 80 ml of ice-cold 10% glycerol, and finally re-suspended in 4 ml of the same buffer. Aliquots of 1 ml were stored frozen at -80°C until use. For electroporation, 50 μl of cells were mixed with 1 μl of DNA and electroporated in 1 mm wide cuvettes (Eurogentec CE-0001-50) at 1660 V using an Eppendorf electroporator 2510. Under these conditions, an average time constant of ~5.4 milliseconds was observed.
Error-prone PCR and Gateway® cloning
Error-prone PCR experiments were performed following the indications provided by the GeneMorph® II Random Mutagenesis Kit instruction manual (Stratagene) using 10 ng of template in 50 μl of PCR mix. At the end of the PCR, 1 μl (20 U) of DpnI (New-England Biolabs) was added to the PCR mix which was then incubated for 1 h at 37°C. After purification, the quality and quantity of the PCR product were estimated as described in the kit manual and by spectrophotometry.
LR reactions were performed overnight at 26°C in a dry incubator in a final volume of 5 μl containing 1.5 μl of each of the donor and acceptor construct, 1 μl of 5× LR buffer, and 1 μl of LR clonase enzyme mix (Invitrogen). The next day, 1 μl of a 2 μg/μl proteinase K solution (Euromedex, EU0090) was added and the reaction mix was incubated for 15 min at 37°C. LR reactions were diluted to 50 μl with water, and 1 μl of this dilution was used to electroporate T7pRos cells as described above. Immediately after electroporation, the cells were resuspended in 15 ml of SOC medium, and then incubated for 1 hour at 37°C under 200 rpm shaking. Serial dilutions of an aliquot were plated on AKCplate to assess the complexity of the library. The remaining bacterial suspension was supplemented with ampicillin, kanamycin and chloramphenicol at a final concentration of 100, 50 and 34 μg/ml, respectively, and grown at 37°C under 200 rpm shaking. When the culture medium became turbid (~5 h), it was supplemented with glycerol at a final concentration of 20% (volume/volume), and 300 μl aliquots were frozen at -80°C.
Library screening
An aliquot of the library was thawed and serial dilutions were spread on AKCplates to assess the number of clones. An aliquot from the same tube was then spread on AKCplates at a cell density allowing isolated colonies to be obtained after overnight growth at 37°C. Colonies were randomly picked up and individually grown at 37°C overnight under 700 rpm shaking in 500 μl of TB containing 100 μg/ml ampicillin, 50 μg/ml kanamycin and 34 μg/ml chloramphenicol (TBAKC) in a 96-well deep-well plate. The next day, another 96-well deep-well plate containing the same volume of the same culture medium was seeded with 50 μl of the overnight culture, and the remaining pre-culture was used to individually seed 96-well ampicillin agar plates provided by GATC Biotech and sent to the same company for plasmid purification and sequencing. The freshly seeded deep-well plate was grown for 1 h at 37°C under 700 rpm shaking. IPTG and arabinose were then added at final concentrations of 0.5 mM and 2%, respectively, and protein expression was allowed to proceed at 17°C for at least 20 h. Culture medium, culture temperature during protein expression, and IPTG and arabinose concentrations leading to the best signal-to-noise ratio were determined by using a fractional factorial approach modified from [28], the detail of which are provided in Additional file 5: Figure S3. At the end of the culture, the deep-well plate was spun for 3 min at 1,500 g and the culture medium discarded. The cell pellets were re-suspended in 500 μl of PBS by shaking at 700 rpm for 20 min at 17°C. The fluorescence of 100 μl of re-suspended cells was measured using a TECAN GENios Plus spectrofluorimeter. The cell density was determined by measuring the optical density at 600 nm (OD600) of 100 μl of re-suspended cells diluted 10 times with PBS, using the same spectrofluorimeter. The results were expressed as the fluorescence to OD600 ratio.
Protein expression and purification
T7pRos cells were extemporaneously co-transformed by heat-shock with the following pairs of constructs: pMRBAD-XD-CGFP and either pNGG-NTAIL (positive control N) or pNGG-Stop-NTAIL (negative control S) or pNGG-NTAIL constructs bearing the mutated NTAIL sequences of interest (clones 1-4); pMRBAD-Z-CGFP and pET11a-Z-NGFP (positive fluorescence control Z). In all cases, transformants were selected on AKCplates. TBAKC (4 ml per well in a 24-well deep-well plate) was seeded with a single colony from AKCplates, and then incubated overnight at 37°C under shaking. The next day, 4 ml of TBAKC in 24-well deep-well plate were seeded with 200 μl of the overnight culture and shaken at 37°C at 200 rpm until the medium became turbid (from ~0.5 to ~1 OD600). IPTG and arabinose were added at final concentrations of 0.5 mM and 2%, respectively, and the cultures were incubated overnight at 17°C under 200 rpm agitation. Cells were then recovered by spinning the deep-well plate for 5 min at 3000 g. Each cell pellet was re-suspended in 1 ml of 50 mM Tris/HCl pH 8, 0.3 M NaCl, 0.1% Triton X100, 1 mM EDTA, 10 mM imidazole, 1 mM PMSF and 0.25 mg/ml lysozyme and frozen. After thawing, DNAseI and MgSO4 were added at final concentrations of 10 μg/ml and 20 mM, respectively and incubated at 37°C in a shaking incubator for 30 min. The deep-well plate was spun for 10 min at 3,000 g at 4°C. The supernatant (soluble fraction) was transferred to 1.5 ml microtubes and supplemented with 50 μl of a 50% (volume/volume) suspension of IMAC sepharose high performance (GE healthcare). The mixture was rotated for one hour on a wheel at 4°C. Sepharose beads were then washed three times with 1 ml of 50 mM Tris/HCl pH 8, 0.3 M NaCl, 50 mM imidazole, and bound His-tagged proteins were eluted with 100 μl of 50 mM Tris/HCl pH 8, 0.3 M NaCl, 500 mM imidazole. When purifying His-tagged proteins under denaturing conditions, the soluble and insoluble fractions were not separated by centrifugation after DNAseI treatment but the total lysate was directly supplemented with two volumes of 50 mM Tris/HCl pH 8, 8 M guanidinium chloride, 0.3 M NaCl, 10 mM imidazole, and with 100 μl of a 50% (volume/volume) suspension of IMAC sepharose beads. Subsequent steps were carried out as described for the non denaturing conditions, except that the experiment was performed at room temperature, and that washing and elution buffers were supplemented with 8 M urea. In all cases, eluted proteins were analyzed by SDS-PAGE.
Abbreviations
- Aa:
-
Amino-acid
- ACplates:
-
LB agar 100 mm plates containing 100 μg/ml ampicillin and 34 μg/ml chloramphenicol
- AKCplates:
-
LB agar 100 mm plates containing 100 μg/ml ampicillin 34 μg/ml chloramphenicol and 50 μg/ml kanamycin
- IPTG:
-
Isopropyl β-D-1-thiogalactopyranoside
- SDS-PAGE:
-
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis
- TB:
-
Turbo broth.
References
-
Vincentelli R, Bignon C, Gruez A, Canaan S, Sulzenbacher G, Tegoni M, Campanacci V, Cambillau C: Medium-scale structural genomics: strategies for protein expression and crystallization. Acc Chem Res 2003,36(3):165-172. 10.1021/ar010130s
Article
CASGoogle Scholar
-
Stemmer WP: DNA shuffling by random fragmentation and reassembly: in vitro recombination for molecular evolution. Proc Natl Acad Sci USA 1994,91(22):10747-10751. 10.1073/pnas.91.22.10747
Article
CASGoogle Scholar
-
Waldo GS, Standish BM, Berendzen J, Terwilliger TC: Rapid protein-folding assay using green fluorescent protein. Nat Biotechnol 1999,17(7):691-695. 10.1038/10904
Article
CASGoogle Scholar
-
van den Berg S, Lofdahl PA, Hard T, Berglund H: Improved solubility of TEV protease by directed evolution. J Biotechnol 2006,121(3):291-298. 10.1016/j.jbiotec.2005.08.006
Article
CASGoogle Scholar
-
Heddle C, Mazaleyrat SL: Development of a screening platform for directed evolution using the reef coral fluorescent protein ZsGreen as a solubility reporter. Protein Eng Des Sel 2007,20(7):327-337. 10.1093/protein/gzm024
Article
CASGoogle Scholar
-
Cao S, Siriwardana CL, Kumimoto RW, Holt BF: Construction of high quality Gateway entry libraries and their application to yeast two-hybrid for the monocot model plant Brachypodium distachyon. BMC Biotechnol 2011, 11: 53. 10.1186/1472-6750-11-53
Article
CASGoogle Scholar
-
Longhi S, Receveur-Brechot V, Karlin D, Johansson K, Darbon H, Bhella D, Yeo R, Finet S, Canard B: The C-terminal domain of the measles virus nucleoprotein is intrinsically disordered and folds upon binding to the C-terminal moiety of the phosphoprotein. J Biol Chem 2003,278(20):18638-18648. 10.1074/jbc.M300518200
Article
CASGoogle Scholar
-
Bourhis J, Johansson K, Receveur-Bréchot V, Oldfield CJ, Dunker AK, Canard B, Longhi S: The C-terminal domain of measles virus nucleoprotein belongs to the class of intrinsically disordered proteins that fold upon binding to their physiological partner. Virus Res 2004, 99: 157-167. 10.1016/j.virusres.2003.11.007
Article
CASGoogle Scholar
-
Johansson K, Bourhis JM, Campanacci V, Cambillau C, Canard B, Longhi S: Crystal structure of the measles virus phosphoprotein domain responsible for the induced folding of the C-terminal domain of the nucleoprotein. J Biol Chem 2003,278(45):44567-44573. 10.1074/jbc.M308745200
Article
CASGoogle Scholar
-
Bourhis JM, Receveur-Bréchot V, Oglesbee M, Zhang X, Buccellato M, Darbon H, Canard B, Finet S, Longhi S: The intrinsically disordered C-terminal domain of the measles virus nucleoprotein interacts with the C-terminal domain of the phosphoprotein via two distinct sites and remains predominantly unfolded. Protein Sci 2005, 14: 1975-1992. 10.1110/ps.051411805
Article
CASGoogle Scholar
-
Morin B, Bourhis JM, Belle V, Woudstra M, Carrière F, BGuigliarelli B, Fournel A, Longhi S: Assessing induced folding of an intrinsically disordered protein by site-directed spin-labeling EPR spectroscopy. J Phys Chem B 2006,110(41):20596-20608. 10.1021/jp063708u
Article
CASGoogle Scholar
-
Belle V, Rouger S, Costanzo S, Liquiere E, Strancar J, Guigliarelli B, Fournel A, Longhi S: Mapping alpha-helical induced folding within the intrinsically disordered C-terminal domain of the measles virus nucleoprotein by site-directed spin-labeling EPR spectroscopy. Proteins: Struct, Funct, Bioinf 2008,73(4):973-988. 10.1002/prot.22125
Article
CASGoogle Scholar
-
Bernard C, Gely S, Bourhis JM, Morelli X, Longhi S, Darbon H: Interaction between the C-terminal domains of N and P proteins of measles virus investigated by NMR. FEBS Lett 2009,583(7):1084-1089. 10.1016/j.febslet.2009.03.004
Article
CASGoogle Scholar
-
Bischak CG, Longhi S, Snead DM, Costanzo S, Terrer E, Londergan CH: Probing structural transitions in the intrinsically disordered C-terminal domain of the measles virus nucleoprotein by vibrational spectroscopy of cyanylated cysteines. Biophys J 2010,99(5):1676-1683. 10.1016/j.bpj.2010.06.060
Article
CASGoogle Scholar
-
Gely S, Lowry DF, Bernard C, Ringkjobing-Jensen M, Blackledge M, Costanzo S, Darbon H, Daughdrill GW, Longhi S: Solution structure of the C-terminal × domain of the measles virus phosphoprotein and interaction with the intrinsically disordered C-terminal domain of the nucleoprotein. J Mol Recognit 2010, 23: 435-447. 10.1002/jmr.1010
Article
CASGoogle Scholar
-
Kavalenka A, Urbancic I, Belle V, Rouger S, Costanzo S, Kure S, Fournel A, Longhi S, Guigliarelli B, Strancar J: Conformational analysis of the partially disordered measles virus NTAIL-XD complex by SDSL EPR spectroscopy. Biophys J 2010,98(6):1055-1064. 10.1016/j.bpj.2009.11.036
Article
CASGoogle Scholar
-
Ringkjøbing Jensen M, Communie G, Ribeiro ED Jr, Martinez N, Desfosses A, Salmon L, Mollica L, Gabel F, Jamin M, Longhi S, et al.: Intrinsic disorder in measles virus nucleocapsids. Proc Natl Acad Sci USA 2011,108(24):9839-9844. 10.1073/pnas.1103270108
Article
Google Scholar
-
Magliery TJ, Wilson CG, Pan W, Mishler D, Ghosh I, Hamilton AD, Regan L: Detecting protein-protein interactions with a green fluorescent protein fragment reassembly trap: scope and mechanism. J Am Chem Soc 2005,127(1):146-157. 10.1021/ja046699g
Article
CASGoogle Scholar
-
Wilson CG, Magliery TJ, Regan L: Detecting protein-protein interactions with GFP-fragment reassembly. Nat Methods 2004,1(3):255-262. 10.1038/nmeth1204-255
Article
CASGoogle Scholar
-
Jackrel ME, Cortajarena AL, Liu TY, Regan L: Screening libraries to identify proteins with desired binding activities using a split-GFP reassembly assay. ACS Chem Biol 2010,5(6):553-562. 10.1021/cb900272j
Article
CASGoogle Scholar
-
Miyazaki K, Takenouchi M: Creating random mutagenesis libraries using megaprimer PCR of whole plasmid. Biotechniques 2002.,33(5): 1033-1034-1036-1038
Google Scholar
-
van den Ent F, Lowe J: RF cloning: a restriction-free method for inserting target genes into plasmids. J Biochem Biophys Methods 2006,67(1):67-74. 10.1016/j.jbbm.2005.12.008
Article
CASGoogle Scholar
-
Fu C, Wehr DR, Edwards J, Hauge B: Rapid one-step recombinational cloning. Nucleic Acids Res 2008,36(9):e54. 10.1093/nar/gkn167
Article
Google Scholar
-
Vincentelli R, Canaan S, Campanacci V, Valencia C, Maurin D, Frassinetti F, Scappucini-Calvo L, Bourne Y, Cambillau C, Bignon C: High-throughput automated refolding screening of inclusion bodies. Protein Sci 2004,13(10):2782-2792.
Article
CASGoogle Scholar
-
Kohli BM, Ostermeier C: A Rubredoxin based system for screening of protein expression conditions and on-line monitoring of the purification process. Protein Expr Purif 2003,28(2):362-367. 10.1016/S1046-5928(02)00704-0
Article
CASGoogle Scholar
-
Stech J, Stech O, Herwig A, Altmeppen H, Hundt J, Gohrbandt S, Kreibich A, Weber S, Klenk HD, Mettenleiter TC: Rapid and reliable universal cloning of influenza A virus genes by target-primed plasmid amplification. Nucleic Acids Res 2008,36(21):e139. 10.1093/nar/gkn646
Article
Google Scholar
-
Karlin D, Longhi S, Canard B: Substitution of two residues in the measles virus nucleoprotein results in an impaired self-association. Virology 2002,302(2):420-432. 10.1006/viro.2002.1634
Article
CASGoogle Scholar
-
Benoit I, Coutard B, Oubelaid R, Asther M, Bignon C: Expression in Escherichia coli, refolding and crystallization of Aspergillus niger feruloyl esterase A using a serial factorial approach. Protein Expr Purif 2007,55(1):166-174. 10.1016/j.pep.2007.04.001
Article
CASGoogle Scholar
Download references
Acknowledgements
The authors wish to thank Dr Lynne Regan for kindly providing pET11a-link-NGFP, pMRBAD-link-CGFP, pET11a-Z-NGFP and pMRBAD-Z-CGFP plasmids, Dr Helena Berglund for the gift of pTH31 and pTH24 expression plasmids, Dr Florence Vincent for LadS and PA3059 encoding plasmids, Dr Bruno Coutard for helpful discussions, and Françoise Delort for reviewing the English manuscript.
This work was funded by the French National Research Agency (ANR) specific programs Physico-Chimie du Vivant (ANR-08-PCVI-0020-01) (S.L.) and E-TRICEL (ANR-07-PNRB-0), and by the European Virus Archive project (FP7 capacities project GA n°228292).
Author information
Authors and Affiliations
-
Architecture et Fonction des Macromolécules Biologiques (AFMB), UMR 7257 CNRS and Aix-Marseille University, 163, Avenue de Luminy, Case 932, Cedex 09, 13288, Marseille, France
Antoine Gruet, Sonia Longhi & Christophe Bignon
Authors
- Antoine Gruet
You can also search for this author in
PubMed Google Scholar - Sonia Longhi
You can also search for this author in
PubMed Google Scholar - Christophe Bignon
You can also search for this author in
PubMed Google Scholar
Corresponding authors
Correspondence to
Sonia Longhi or Christophe Bignon.
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
AG obtained the original plasmids from Dr.LR, performed most of the experiments and participated in the writing of the manuscript. SL had the idea of using directed evolution for studying NTAIL-XD interaction, is the principal investigator of the ANR-08-PCVI-0020-01 program and participated in the writing of the manuscript. CB devised the method, participated in some experiments, directed the work of the first author and wrote the paper. All authors read and approved the final manuscript.
Electronic supplementary material
Authors’ original submitted files for images
Rights and permissions
Open Access
This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License (
https://creativecommons.org/licenses/by/2.0
), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Reprints and Permissions
About this article
Cite this article
Gruet, A., Longhi, S. & Bignon, C. One-step generation of error-prone PCR libraries using Gateway® technology.
Microb Cell Fact 11, 14 (2012). https://doi.org/10.1186/1475-2859-11-14
Download citation
-
Received: 08 November 2011
-
Accepted: 30 January 2012
-
Published: 30 January 2012
-
DOI: https://doi.org/10.1186/1475-2859-11-14
Keywords
- Cloning
- Sub-cloning
- Gateway®
- Directed evolution
- GFP
- Error-prone PCR
- epPCR
- Library
- Screening
Error prone PCR is one of the methods for random mutagenesis in vitro. The main idea is to use non-high-fidelity DNA polymerase
DNA polymerase
The DNA polymerases are enzymes that create DNA molecules by assembling nucleotides, the building blocks of DNA. These enzymes are essential to DNA replication and usually work in pairs to create two identical DNA strands from a single original DNA molecule. During this proces…
, such as Taq, and introduce ‘sloppy’ reaction conditions, e.g. by adding Mg2+ or Mn2+. Under these these ‘sloppy’ conditions Taq will introduce even more errors during synthesis.
Error-prone PCR (EP-PCR) is the method of choice for introducing random mutations into a defined segment of DNA that is too long to be chemically synthesized as a degenerate sequence (UNIT 8.2A). The 5′ and 3′ boundaries of the mutated region may be defined by the choice of PCR primers.
Full
Answer
What is the error prone PCR method?
Error prone PCR is one of the methods for random mutagenesis in vitro. The main idea is to use non-high-fidelity DNA polymerase, such as Taq, and introduce ‘sloppy’ reaction conditions, e.g. by adding Mg2+ or Mn2+. Under these these ‘sloppy’ conditions Taq will introduce even more errors during synthesis.
Is error-prone PCR less effective at generating grossly divergent sequences?
Error-prone PCR in realistic reaction conditions is predicted to be less effective at generating grossly divergent sequences than the original model. The estimate of mutation rate per cycle by sampling sequences from an in vitro PCR experiment is correspondingly affected by the choice of model and parameters.
What is random mutagenesis by PCR?
Random mutagenesis by PCR Error-prone PCR (EP-PCR) is the method of choice for introducing random mutations into a defined segment of DNA that is too long to be chemically synthesized as a degenerate sequence. Using EP-PCR, the 5′ and 3′ boundaries of the mutated region may be defined by the choice of PCR primers.
How do I perform error-prone PCR experiments with the genemorph II?
Error-prone PCR experiments were performed following the indications provided by the GeneMorph ® II Random Mutagenesis Kit instruction manual (Stratagene) using 10 ng of template in 50 μl of PCR mix. At the end of the PCR, 1 μl (20 U) of DpnI (New-England Biolabs) was added to the PCR mix which was then incubated for 1 h at 37°C.
What is an error prone polymerase?
Error-prone DNA polymerases are characterized by probabilities of base substitution or frameshift mutations ranging from 10−3 to 7.5 · 10−1 in an intact DNA, whereas the spontaneous mutagenesis rate per replicated nucleotide varies between 10−10 and 10−12.
Which polymerase is used in error prone PCR?
Mutazyme II DNA polymeraseMutazyme II DNA polymerase is a novel error prone PCR enzyme blend, formulated to provide useful mutation rates with minimal mutational bias.
What are the most common problems encountered when designing PCR primers?
There are a few common problems that arise when designing primers: 1) self-annealing of primers resulting in formation of secondary structures such as hairpin loops (Figure 1a); 2) primer annealing to each other, rather then the DNA template, creating primer dimers (Figure 1b); 3) drastically different melting …
Error-prone PCR protocols are modifications of standard PCR methods, designed to alter and enhance the natural error rate of the polymerase (1,2). Taq polymerase (3) is commonly used because of its naturally high error rate, with errors biased toward AT to GC changes.
What does error prone mean?
Adjective. error-prone (comparative more error-prone, superlative most error-prone) In the habit of making errors.
What is the error prone PCR and why it is useful?
Error-prone PCR (EP-PCR) is the method of choice for introducing random mutations into a defined segment of DNA that is too long to be chemically synthesized as a degenerate sequence. Using EP-PCR, the 5′ and 3′ boundaries of the mutated region may be defined by the choice of PCR primers.
What causes PCR failure?
Forgetting just one component of the PCR reaction, whether that be the DNA polymerase, primers or even the template DNA, will result in a failed reaction. The simplest solution is to repeat the reaction. Take your time to ensure everything has been added.
What happens if only one primer is used in PCR?
If only one primer is used, the process is called “asymmetric PCR”. Only one strand of the double-stranded DNA will be amplified, and only one new copy is synthesized per cycle, which is unable to achieve exponential amplification.
What happens if you add too much primer to a PCR?
Using an excessive concentration of primers can increase the chance of primers binding nonspecifically to undesired sites on the template or to each other. Use well-designed primers at 0.2–1 μM in the final reaction.
How can error prone PCR be run?
Error-prone PCR (EP-PCR) is the method of choice for introducing random mutations into a defined segment of DNA that is too long to be chemically synthesized as a degenerate sequence (UNIT 8.2A)….STRATEGIC PLANNING.Type of mutationNumber times observedA->C and T->G6G->C and C->G5G->T and C->A23 more rows
Which of these enzymes is most error prone?
Thus, polι is highly error-prone compared to most replicative enzymes and even appears to have slightly lower fidelity than that recently reported for the related human polη (Matsuda et al.
What is the error rate of Taq polymerase?
For Taq polymerase, which has a total error rate of 1.8 × 10−4 errors/base/doubling, single-molecule sequencing was sufficient to detect all types of polymerase errors, including base substitutions, deletions and insertions.
What is error prone PCR?
Error-prone PCR (epPCR) is a popular technique employed in synthetic biology and directed evolution to incorporate mutations across a wide genetic background, typically entire protein coding sequences (CDS) [ [1], [2], [3], [4] ] . Compared to cassette mutagenesis (also called synthetic degenerate oligonucleotide PCR), which utilizes standard PCR conditions but with degenerate pools of oligonucleotide primers, epPCR takes advantage of the imperfect fidelity of DNA polymerase to periodically misincorporate a non-complementary dNTP during template-mediated elongation. The result is a mutation on the newly synthesized daughter strand that is then propagated to half of the double-stranded progeny upon further replication cycles. The usefulness (and enduring popularity) of epPCR stems from the fact that it enables a broad mutational spectrum and can be particularly useful when the experimenter does not know a priori which amino acid positions in a protein one would like to target for mutagenesis [ 5 ]. It can also be useful when the property which one would like to select for is not determined by any single region of the protein of interest (e.g., thermostability) [ 4, 6 ]. In the typical protocol, the CDS targeted for mutagenesis is amplified under error-prone conditions, whilst the backbone region of the vector (including its selectable marker and origin of replication) is amplified by high-fidelity PCR. The insert, encoding a combinatorial library, is then ligated into a constant vector, resulting in a plasmid library.
Which polymerases have the advantage of generating less biased mutational spectra?
Though nucleotide analogs and mutagenic buffers can lead to even higher error rates (approaching 1 in 10 2 nucleotides), mutant polymerases have the advantage of generating less biased mutational spectra [ 1 ].
Why is error prone PCR useful?
Error-prone PCR may be used deliberately to generate random mutations , but the potential for generating random mutations is always present in any PCR experiment. In either case it is useful to be able to predict and understand the distribution and frequency of occurrence of random mutations. When deliberately generating random mutations to explore sequence space it is helpful to know the size of sequence space that may potentially be explored by the generated mutations; estimates of mutation rate per cycle and an understanding of how to maximize overall mutation rate per sequence are important to this aim. In the case of amplifying or proof-reading PCR, where mutations are undesirable, it is useful to know how many random mutations are likely to occur, and how best to minimize them. Again, an estimate of the mutation rate per cycle and an understanding of how to minimise overall mutation rate will be important. If the mutation rate is constant for each generation, it is expected that the number of mutations observed in a single sequence extracted from the final pool will be proportional to the number of extension steps of which it is a product. What is important is how many sequences from each generation are present in the final pool. This is represented by , which denotes how many sequences present in the pot after a total of N cycles are the result of n extension steps. Control of the modal number of extension steps may therefore be an important mechanism for determining overall mutation rate.
What is PCR in biology?
The polymerase chain reaction (PCR) is an in vitro method capable of producing large amounts of identical copies of a gene or other specified nucleotide sequence from a small amount of DNA ( Erlich, 1989, Mullis and Faloona, 1987, Sun, 1995 ). PCR is essentially a three-stage process that amplifies the region of DNA lying between two short …
What is the initial mutation data matrix?
The protocol of the model proposed by Moore and Maranas (2000) may be summarised as follows:#N#1.#N#An initial mutation data matrix is defined representing nucleotide-dependent substitutions for a single cycle of the PCR experiment: (1) Here, represents the probability that nucleotide i will be substituted by nucleotide j in a single PCR cycle.#N#2.#N#Mutation data matrices for successive cycles of the PCR experiment are generated (this is assumed to be independent of other factors, such as the uneven depletion of nucleotides that may have a large effect in real PCR). (2) where is the Kronecker delta.#N#3.#N#The number of sequences produced at each cycle n of a PCR experiment that runs for a total of N cycles is calculated as . These values will usually be dependent on the amplification efficiency, which itself may be variable and dependent on the initial population size. For an arbitrary but constant amplification efficiency , and arbitrary initial number of template sequences , after N total PCR cycles the number of sequences present that are the result of n extension steps is given by (3) as suggested in Moore and Maranas (2000) (proof in Appendix B). The total number of sequences at the end of the experiment is given by (see Appendix A) (4) For an arbitrary amplification efficiency that is also dependent on the current PCR cycle, we performed the recursive procedure explicitly. The analytical solution of the total number of sequences at the end of the experiment is given by (Appendix C) (5) The total number of sequences generated by an in silico experiment as described here, and the number of sequences generated after n cycles of such an experiment, are determined by these equations. The only influence of stochasticity on these models is derived entirely from the substitution process. The assumption is made throughout these models that mutations at different locations along the sequence are independent. This is probably true if, as is the case with in vitro selection, there is no selective pressure between rounds of screening. As such, a sum of the product of the mutation data matrix for each generation and the proportion of all sequences in the final PCR pot contributed by that generation will produce an aggregate mutation data matrix representing the probabilities of any base having mutated by cycle N, represented by . (6)#N#4.#N#The application of this aggregate mutation data matrix to each base in a given template sequence generates a sequence that is equivalent to the random selection of a single sequence from the final PCR pot.
What is error prone PCR?
Error-prone PCR (epPCR) libraries are one of the tools used in directed evolution. The Gateway ® technology allows constructing epPCR libraries virtually devoid of any background ( i.e ., of insert-free plasmid), but requires two steps: the BP and the LR reactions and the associated E. coli cell transformations and plasmid purifications.
How many base pairs are needed for RF cloning?
In contrast to MEGAWHOP, not the whole length of the linear DNA to clone is used for hybridization, but only 24 base pairs at both ends which are designed to be complementary to the cloning site of the plasmid.
What is a pMRBAD link?
pMRBAD-link-CGFP [ 19 ], encoding the C-terminal half (aa 158-238) of eGFP under the control of the P BAD promoter (inducible by L-arabinose) and allowing upstream in frame cloning of DNA fragments, was a kind gift of Lynne Regan, as were pET11a-link-NGFP, pET11a-Z-NGFP and pMRBAD-Z-CGFP. These latter two encode fusion proteins between leucine zippers and the two halves of the eGFP, while pET11a-link-NGFP encodes the N-terminal half (aa 1-157) of eGFP under the control of the T7 promoter (inducible by IPTG) and allows downstream in-frame cloning of DNA fragments [ 19 ]. pMRBAD-XD-CGFP was constructed as follows. The coding sequence of XD was PCR amplified using primers Nco1-MeV.XD and MeV.XD-AatII (Table 6) and a plasmid encoding the X domain (aa 459-507) of the measles virus phosphoprotein (strain Edmonston B) (pDEST14/XD HC) as template [ 9 ]. DpnI-treated purified PCR product and pMRBAD-link-CGFP were ligated after NcoI and AatII digestion. pDONR-N TAIL was obtained by BP reaction with a PCR N TAIL fragment amplified using primers N TAIL F and N TAIL R (Table 6 ), and a plasmid encoding the measles virus nucleoprotein (strain Edmonston B) (pET-21a/N) as template [ 27 ]. pDONR-Stop-N TAIL was obtained by BP reaction with a PCR N TAIL fragment amplified using primers StopNtail and N TAIL R (Table 6) and pDONR-N TAIL as template. StopNtail has two Stop codons before the first N TAIL codon. pNGG-N TAIL and pNGG-Stop-N TAIL were obtained by LR reaction using pNGG as destination vector and pDONR-N TAIL or pDONR-Stop-N TAIL as donor constructs, respectively. pDEST17O/I-N TAIL was obtained by LR reaction using pDONR-N TAIL and pDEST17O/I [ 24 ]. The constructs were verified by sequencing and found to conform to expectations. The plasmids used in this study are summarized in Table 2.
Popular Answers (1)
Error prone PCR is one of the methods for random mutagenesis in vitro. The main idea is to use non-high-fidelity DNA polymerase, such as Taq, and introduce ‘sloppy’ reaction conditions, e.g. by adding Mg2+ or Mn2+. Under these these ‘sloppy’ conditions Taq will introduce even more errors during synthesis.
All Answers (3)
Error prone PCR is one of the methods for random mutagenesis in vitro. The main idea is to use non-high-fidelity DNA polymerase, such as Taq, and introduce ‘sloppy’ reaction conditions, e.g. by adding Mg2+ or Mn2+. Under these these ‘sloppy’ conditions Taq will introduce even more errors during synthesis.
If no amplification products are obtained, what parameters should be considered first when troubleshooting?
First, ensure that all PCR components were included in the reactions. A positive control should always be included to ensure that each component is present and functional.
If PCR generates a smear after running the products on a gel, what can be done to improve the results?
First, determine the source of the smear using positive and negative (no template) controls. This can determine if the cause of the smear is contamination or overcycling, or if the smear results from poorly designed primers or suboptimal PCR conditions.
What are some sources of PCR contamination?
The most common source of contamination is PCR product from previous amplifications (called «carryover contamination»). When large amounts of PCR product (10 12 molecules) are generated repeatedly over a period of time, the potential for contamination increases.
How can contamination be avoided?
The sensitivity of PCR requires that samples are not contaminated with any exogenous DNA or any previously amplified products from the laboratory environment. We recommend that distinct areas are used for sample preparation, PCR setup, and post-PCR analysis.
How can I decontaminate if I have PCR contamination?
Leave pipettes under UV light in the cell culture hood overnight. UV irradiation promotes cross-linking of thymidine residues, damaging residual DNA.
What can I do if the PCR generates errors?
To avoid errors during PCR, we recommend using a high-fidelity enzyme ( see selection guide ). In addition, be sure to avoid the following:
What are PCR inhibitors?
Impurities that interfere with PCR amplification are known as PCR inhibitors. PCR inhibitors are present in a large variety of sample types and may lead to decreased PCR sensitivity or even false-negative PCR results. PCR inhibitors may have both inorganic and organic origins (Schrader 2012).
Generation of An epPCR Library
The conventional procedure for generating epPCR libraries using the Gateway® technology comprises two recombination reactions (BP and LR) [4]. We first addressed the question as to whether each recombination reaction and associated E. coli cell transformation decreased the complexity of a given library. A typical G…
See more on microbialcellfactories.biomedcentral.com
Sub-Cloning of Selected Mutated Sequences
-
After mutants of interest have been selected, their coding sequence must generally be sub-cloned from the reporter expression plasmid used to screen the epPCR library to another non-reporter expression plasmid (Figure 1, stage 2). In the regular strategy [4], this procedure essentially follows the same process as that used to create the random library (compare stage 1 and stage …
See more on microbialcellfactories.biomedcentral.com
Application of The Method to The Ntail-Xd Interaction Project
-
We have applied the above-described strategy to few directed evolution projects (B. Coutard, F. Vincent, unpublished data), including the one reported herein. In view of gaining insights into the NTAIL-XD interaction, we generated a mutant library of NTAIL and used a split GFP re-assembly assay to screen variants with altered interaction abilities [18–20]. In this method, the sequence c…
See more on microbialcellfactories.biomedcentral.com
Popular Posts:
Random mutagenesis by PCR
D S Wilson et al.
Curr Protoc Mol Biol.
2001 May.
Abstract
Error-prone PCR (EP-PCR) is the method of choice for introducing random mutations into a defined segment of DNA that is too long to be chemically synthesized as a degenerate sequence. Using EP-PCR, the 5′ and 3′ boundaries of the mutated region may be defined by the choice of PCR primers. Accordingly, it is possible to mutagenize an entire gene or merely a segment of a gene. The average number of mutations per DNA fragment can be controlled as a function of the number of EP-PCR doublings performed. The EP-PCR technique described here is for a 400-bp sequence, and an Alternate Protocol is for a library. EP-PCR takes advantage of the inherently low fidelity of Taq DNA polymerase, which may be further decreased by the addition of Mn2+, increasing the Mg2+ concentration, and using unequal dNTP concentrations.
Similar articles
-
Thermostable DNA ligase-mediated PCR production of circular plasmid (PPCP) and its application in directed evolution via in situ error-prone PCR.
Le Y, Chen H, Zagursky R, Wu JH, Shao W.
Le Y, et al.
DNA Res. 2013 Aug;20(4):375-82. doi: 10.1093/dnares/dst016. Epub 2013 Apr 30.
DNA Res. 2013.PMID: 23633530
Free PMC article. -
Novel PCR-mediated mutagenesis employing DNA containing a natural abasic site as a template and translesional Taq DNA polymerase.
Kobayashi A, Kitaoka M, Hayashi K.
Kobayashi A, et al.
J Biotechnol. 2005 Mar 30;116(3):227-32. doi: 10.1016/j.jbiotec.2004.10.016. Epub 2004 Dec 25.
J Biotechnol. 2005.PMID: 15707683
-
An efficient and optimized PCR method with high fidelity for site-directed mutagenesis.
Liang Q, Chen L, Fulco AJ.
Liang Q, et al.
PCR Methods Appl. 1995 Apr;4(5):269-74. doi: 10.1101/gr.4.5.269.
PCR Methods Appl. 1995.PMID: 7580913
-
Critical evaluation of random mutagenesis by error-prone polymerase chain reaction protocols, Escherichia coli mutator strain, and hydroxylamine treatment.
Rasila TS, Pajunen MI, Savilahti H.
Rasila TS, et al.
Anal Biochem. 2009 May 1;388(1):71-80. doi: 10.1016/j.ab.2009.02.008. Epub 2009 Feb 10.
Anal Biochem. 2009.PMID: 19454214
-
[Polymerase chain reaction, cold probes and clinical diagnosis].
Haras D, Amoros JP.
Haras D, et al.
Sante. 1994 Jan-Feb;4(1):43-52.
Sante. 1994.PMID: 7909267
Review.
French.
Cited by
-
RNA-controlled nucleocytoplasmic shuttling of mRNA decay factors regulates mRNA synthesis and a novel mRNA decay pathway.
Chattopadhyay S, Garcia-Martinez J, Haimovich G, Fischer J, Khwaja A, Barkai O, Chuartzman SG, Schuldiner M, Elran R, Rosenberg MI, Urim S, Deshmukh S, Bohnsack KE, Bohnsack MT, Perez-Ortin JE, Choder M.
Chattopadhyay S, et al.
Nat Commun. 2022 Nov 23;13(1):7184. doi: 10.1038/s41467-022-34417-z.
Nat Commun. 2022.PMID: 36418294
Free PMC article. -
Mutagenesis and structural modeling implicate RME-8 IWN domains as conformational control points.
Norris A, McManus CT, Wang S, Ying R, Grant BD.
Norris A, et al.
PLoS Genet. 2022 Oct 24;18(10):e1010296. doi: 10.1371/journal.pgen.1010296. eCollection 2022 Oct.
PLoS Genet. 2022.PMID: 36279308
Free PMC article. -
Generation and characterization of conditional yeast mutants affecting each of the 2 essential functions of the scaffolding proteins Boi1/2 and Bem1.
Sulpizio A, Herpin L, Gingras R, Liu W, Bretscher A.
Sulpizio A, et al.
G3 (Bethesda). 2022 Dec 1;12(12):jkac273. doi: 10.1093/g3journal/jkac273.
G3 (Bethesda). 2022.PMID: 36218417
Free PMC article. -
Sir2 and Reb1 antagonistically regulate nucleosome occupancy in subtelomeric X-elements and repress TERRAs by distinct mechanisms.
Bauer SL, Grochalski TNT, Smialowska A, Åström SU.
Bauer SL, et al.
PLoS Genet. 2022 Sep 22;18(9):e1010419. doi: 10.1371/journal.pgen.1010419. eCollection 2022 Sep.
PLoS Genet. 2022.PMID: 36137093
Free PMC article. -
Phosphate Mobilization by Culturable Fungi and Their Capacity to Increase Soil P Availability and Promote Barley Growth.
Brazhnikova YV, Shaposhnikov AI, Sazanova AL, Belimov AA, Mukasheva TD, Ignatova LV.
Brazhnikova YV, et al.
Curr Microbiol. 2022 Jul 6;79(8):240. doi: 10.1007/s00284-022-02926-1.
Curr Microbiol. 2022.PMID: 35792979
MeSH terms
Substances
LinkOut — more resources
-
Full Text Sources
- Wiley
-
Other Literature Sources
- The Lens — Patent Citations
Protein engineering is the process of developing useful or valuable protein and design and production of unnatural polypeptides, often by altering amino acid sequences found in nature.[1] It is a young discipline, with much research taking place into the understanding of protein folding and recognition for protein design principles. It has been used to improve the function of many enzymes for industrial catalysis.[2] It is also a product and services market, with an estimated value of $168 billion by 2017.[3]
There are two general strategies for protein engineering: rational protein design and directed evolution. These methods are not mutually exclusive; researchers will often apply both. In the future, more detailed knowledge of protein structure and function, and advances in high-throughput screening, may greatly expand the abilities of protein engineering. Eventually, even unnatural amino acids may be included, via newer methods, such as expanded genetic code, that allow encoding novel amino acids in genetic code.
Approaches[edit]
Rational design[edit]
In rational protein design, a scientist uses detailed knowledge of the structure and function of a protein to make desired changes. In general, this has the advantage of being inexpensive and technically easy, since site-directed mutagenesis methods are well-developed. However, its major drawback is that detailed structural knowledge of a protein is often unavailable, and, even when available, it can be very difficult to predict the effects of various mutations since structural information most often provide a static picture of a protein structure. However, programs such as Folding@home and Foldit have utilized crowdsourcing techniques in order to gain insight into the folding motifs of proteins.[4]
Computational protein design algorithms seek to identify novel amino acid sequences that are low in energy when folded to the pre-specified target structure. While the sequence-conformation space that needs to be searched is large, the most challenging requirement for computational protein design is a fast, yet accurate, energy function that can distinguish optimal sequences from similar suboptimal ones.
Multiple sequence alignment[edit]
Without structural information about a protein, sequence analysis is often useful in elucidating information about the protein. These techniques involve alignment of target protein sequences with other related protein sequences. This alignment can show which amino acids are conserved between species and are important for the function of the protein. These analyses can help to identify hot spot amino acids that can serve as the target sites for mutations. Multiple sequence alignment utilizes data bases such as PREFAB, SABMARK, OXBENCH, IRMBASE, and BALIBASE in order to cross reference target protein sequences with known sequences. Multiple sequence alignment techniques are listed below.[5][page needed]
This method begins by performing pair wise alignment of sequences using k-tuple or Needleman–Wunsch methods. These methods calculate a matrix that depicts the pair wise similarity among the sequence pairs. Similarity scores are then transformed into distance scores that are used to produce a guide tree using the neighbor joining method. This guide tree is then employed to yield a multiple sequence alignment.[5][page needed]
Clustal omega[edit]
This method is capable of aligning up to 190,000 sequences by utilizing the k-tuple method. Next sequences are clustered using the mBed and k-means methods. A guide tree is then constructed using the UPGMA method that is used by the HH align package. This guide tree is used to generate multiple sequence alignments.[5][page needed]
MAFFT[edit]
This method utilizes fast Fourier transform (FFT) that converts amino acid sequences into a sequence composed of volume and polarity values for each amino acid residue. This new sequence is used to find homologous regions.[5][page needed]
K-Align[edit]
This method utilizes the Wu-Manber approximate string matching algorithm to generate multiple sequence alignments.[5][page needed]
Multiple sequence comparison by log expectation (MUSCLE)[edit]
This method utilizes Kmer and Kimura distances to generate multiple sequence alignments.[5][page needed]
T-Coffee[edit]
This method utilizes tree based consistency objective functions for alignment evolution. This method has been shown to be 5-10% more accurate than Clustal W.[5][page needed]
Coevolutionary analysis[edit]
Coevolutionary analysis is also known as correlated mutation, covariation, or co-substitution. This type of rational design involves reciprocal evolutionary changes at evolutionarily interacting loci. Generally this method begins with the generation of a curated multiple sequence alignments for the target sequence. This alignment is then subjected to manual refinement that involves removal of highly gapped sequences, as well as sequences with low sequence identity. This step increases the quality of the alignment. Next, the manually processed alignment is utilized for further coevolutionary measurements using distinct correlated mutation algorithms. These algorithms result in a coevolution scoring matrix. This matrix is filtered by applying various significance tests to extract significant coevolution values and wipe out background noise. Coevolutionary measurements are further evaluated to assess their performance and stringency. Finally, the results from this coevolutionary analysis are validated experimentally.[5][page needed]
Structural prediction[edit]
De novo synthesis of protein benefits from knowledge of existing protein structures. This knowledge of existing protein structure assists with the prediction of new protein structures. Methods for protein structure prediction fall under one of the four following classes: ab initio, fragment based methods, homology modeling, and protein threading.[5][page needed]
Ab initio[edit]
These methods involve free modeling without using any structural information about the template. Ab initio methods are aimed at prediction of the native structures of proteins corresponding to the global minimum of its free energy. some examples of ab initio methods are AMBER, GROMOS, GROMACS, CHARMM, OPLS, and ENCEPP12. General steps for ab initio methods begin with the geometric representation of the protein of interest. Next, a potential energy function model for the protein is developed. This model can be created using either molecular mechanics potentials or protein structure derived potential functions. Following the development of a potential model, energy search techniques including molecular dynamic simulations, Monte Carlo simulations and genetic algorithms are applied to the protein.[5][page needed]
Fragment based[edit]
These methods use database information regarding structures to match homologous structures to the created protein sequences. These homologous structures are assembled to give compact structures using scoring and optimization procedures, with the goal of achieving the lowest potential energy score. Webservers for fragment information are I-TASSER, ROSETTA, ROSETTA @ home, FRAGFOLD, CABS fold, PROFESY, CREF, QUARK, UNDERTAKER, HMM, and ANGLOR.[5]: 72
Homology modeling[edit]
These methods are based upon the homology of proteins. These methods are also known as comparative modeling. The first step in homology modeling is generally the identification of template sequences of known structure which are homologous to the query sequence. Next the query sequence is aligned to the template sequence. Following the alignment, the structurally conserved regions are modeled using the template structure. This is followed by the modeling of side chains and loops that are distinct from the template. Finally the modeled structure undergoes refinement and assessment of quality. Servers that are available for homology modeling data are listed here: SWISS MODEL, MODELLER, ReformAlign, PyMOD, TIP-STRUCTFAST, COMPASS, 3d-PSSM, SAMT02, SAMT99, HHPRED, FAGUE, 3D-JIGSAW, META-PP, ROSETTA, and I-TASSER.[5][page needed]
Protein threading[edit]
Protein threading can be used when a reliable homologue for the query sequence cannot be found. This method begins by obtaining a query sequence and a library of template structures. Next, the query sequence is threaded over known template structures. These candidate models are scored using scoring functions. These are scored based upon potential energy models of both query and template sequence. The match with the lowest potential energy model is then selected. Methods and servers for retrieving threading data and performing calculations are listed here: GenTHREADER, pGenTHREADER, pDomTHREADER, ORFEUS, PROSPECT, BioShell-Threading, FFASO3, RaptorX, HHPred, LOOPP server, Sparks-X, SEGMER, THREADER2, ESYPRED3D, LIBRA, TOPITS, RAPTOR, COTH, MUSTER.[5][page needed]
For more information on rational design see site-directed mutagenesis.
Multivalent binding[edit]
Multivalent binding can be used to increase the binding specificity and affinity through avidity effects. Having multiple binding domains in a single biomolecule or complex increases the likelihood of other interactions to occur via individual binding events. Avidity or effective affinity can be much higher than the sum of the individual affinities providing a cost and time-effective tool for targeted binding.[6]
Multivalent proteins[edit]
Multivalent proteins are relatively easy to produce by post-translational modifications or multiplying the protein-coding DNA sequence. The main advantage of multivalent and multispecific proteins is that they can increase the effective affinity for a target of a known protein. In the case of an inhomogeneous target using a combination of proteins resulting in multispecific binding can increase specificity, which has high applicability in protein therapeutics.
The most common example for multivalent binding are the antibodies, and there is extensive research for bispecific antibodies. Applications of bispecific antibodies cover a broad spectrum that includes diagnosis, imaging, prophylaxis, and therapy.[7][8]
Directed evolution[edit]
In directed evolution, random mutagenesis, e.g. by error-prone PCR or sequence saturation mutagenesis, is applied to a protein, and a selection regime is used to select variants having desired traits. Further rounds of mutation and selection are then applied. This method mimics natural evolution and, in general, produces superior results to rational design. An added process, termed DNA shuffling, mixes and matches pieces of successful variants to produce better results. Such processes mimic the recombination that occurs naturally during sexual reproduction. Advantages of directed evolution are that it requires no prior structural knowledge of a protein, nor is it necessary to be able to predict what effect a given mutation will have. Indeed, the results of directed evolution experiments are often surprising in that desired changes are often caused by mutations that were not expected to have some effect. The drawback is that they require high-throughput screening, which is not feasible for all proteins. Large amounts of recombinant DNA must be mutated and the products screened for desired traits. The large number of variants often requires expensive robotic equipment to automate the process. Further, not all desired activities can be screened for easily.
Natural Darwinian evolution can be effectively imitated in the lab toward tailoring protein properties for diverse applications, including catalysis. Many experimental technologies exist to produce large and diverse protein libraries and for screening or selecting folded, functional variants. Folded proteins arise surprisingly frequently in random sequence space, an occurrence exploitable in evolving selective binders and catalysts. While more conservative than direct selection from deep sequence space, redesign of existing proteins by random mutagenesis and selection/screening is a particularly robust method for optimizing or altering extant properties. It also represents an excellent starting point for achieving more ambitious engineering goals. Allying experimental evolution with modern computational methods is likely the broadest, most fruitful strategy for generating functional macromolecules unknown to nature.[9]
The main challenges of designing high quality mutant libraries have shown significant progress in the recent past. This progress has been in the form of better descriptions of the effects of mutational loads on protein traits. Also computational approaches have showed large advances in the innumerably large sequence space to more manageable screenable sizes, thus creating smart libraries of mutants. Library size has also been reduced to more screenable sizes by the identification of key beneficial residues using algorithms for systematic recombination. Finally a significant step forward toward efficient reengineering of enzymes has been made with the development of more accurate statistical models and algorithms quantifying and predicting coupled mutational effects on protein functions.[10]
Generally, directed evolution may be summarized as an iterative two step process which involves generation of protein mutant libraries, and high throughput screening processes to select for variants with improved traits. This technique does not require prior knowledge of the protein structure and function relationship. Directed evolution utilizes random or focused mutagenesis to generate libraries of mutant proteins. Random mutations can be introduced using either error prone PCR, or site saturation mutagenesis. Mutants may also be generated using recombination of multiple homologous genes. Nature has evolved a limited number of beneficial sequences. Directed evolution makes it possible to identify undiscovered protein sequences which have novel functions. This ability is contingent on the proteins ability to tolerant amino acid residue substitutions without compromising folding or stability.[5][page needed]
Directed evolution methods can be broadly categorized into two strategies, asexual and sexual methods.
Asexual methods[edit]
Asexual methods do not generate any cross links between parental genes. Single genes are used to create mutant libraries using various mutagenic techniques. These asexual methods can produce either random or focused mutagenesis.
Random mutagenesis[edit]
Random mutagenic methods produce mutations at random throughout the gene of interest. Random mutagenesis can introduce the following types of mutations: transitions, transversions, insertions, deletions, inversion, missense, and nonsense. Examples of methods for producing random mutagenesis are below.
Error prone PCR[edit]
Error prone PCR utilizes the fact that Taq DNA polymerase lacks 3′ to 5′ exonuclease activity. This results in an error rate of 0.001-0.002% per nucleotide per replication. This method begins with choosing the gene, or the area within a gene, one wishes to mutate. Next, the extent of error required is calculated based upon the type and extent of activity one wishes to generate. This extent of error determines the error prone PCR strategy to be employed. Following PCR, the genes are cloned into a plasmid and introduced to competent cell systems. These cells are then screened for desired traits. Plasmids are then isolated for colonies which show improved traits, and are then used as templates the next round of mutagenesis. Error prone PCR shows biases for certain mutations relative to others. Such as biases for transitions over transversions.[5][page needed]
Rates of error in PCR can be increased in the following ways:[5][page needed]
- Increase concentration of magnesium chloride, which stabilizes non complementary base pairing.
- Add manganese chloride to reduce base pair specificity.
- Increased and unbalanced addition of dNTPs.
- Addition of base analogs like dITP, 8 oxo-dGTP, and dPTP.
- Increase concentration of Taq polymerase.
- Increase extension time.
- Increase cycle time.
- Use less accurate Taq polymerase.
Also see polymerase chain reaction for more information.
Rolling circle error-prone PCR[edit]
This PCR method is based upon rolling circle amplification, which is modeled from the method that bacteria use to amplify circular DNA. This method results in linear DNA duplexes. These fragments contain tandem repeats of circular DNA called concatamers, which can be transformed into bacterial strains. Mutations are introduced by first cloning the target sequence into an appropriate plasmid. Next, the amplification process begins using random hexamer primers and Φ29 DNA polymerase under error prone rolling circle amplification conditions. Additional conditions to produce error prone rolling circle amplification are 1.5 pM of template DNA, 1.5 mM MnCl2 and a 24 hour reaction time. MnCl2 is added into the reaction mixture to promote random point mutations in the DNA strands. Mutation rates can be increased by increasing the concentration of MnCl2, or by decreasing concentration of the template DNA. Error prone rolling circle amplification is advantageous relative to error prone PCR because of its use of universal random hexamer primers, rather than specific primers. Also the reaction products of this amplification do not need to be treated with ligases or endonucleases. This reaction is isothermal.[5][page needed]
Chemical mutagenesis[edit]
Chemical mutagenesis involves the use of chemical agents to introduce mutations into genetic sequences. Examples of chemical mutagens follow.
Sodium bisulfate is effective at mutating G/C rich genomic sequences. This is because sodium bisulfate catalyses deamination of unmethylated cytosine to uracil.[5][page needed]
Ethyl methane sulfonate alkylates guanidine residues. This alteration causes errors during DNA replication.[5][page needed]
Nitrous acid causes transversion by de-amination of adenine and cytosine.[5][page needed]
The dual approach to random chemical mutagenesis is an iterative two step process. First it involves the in vivo chemical mutagenesis of the gene of interest via EMS. Next, the treated gene is isolated and cloning into an untreated expression vector in order to prevent mutations in the plasmid backbone.[5][page needed] This technique preserves the plasmids genetic properties.[5][page needed]
Targeting glycosylases to embedded arrays for mutagenesis (TaGTEAM)[edit]
This method has been used to create targeted in vivo mutagenesis in yeast. This method involves the fusion of a 3-methyladenine DNA glycosylase to tetR DNA-binding domain. This has been shown to increase mutation rates by over 800 time in regions of the genome containing tetO sites.[5][page needed]
Mutagenesis by random insertion and deletion[edit]
This method involves alteration in length of the sequence via simultaneous deletion and insertion of chunks of bases of arbitrary length. This method has been shown to produce proteins with new functionalities via introduction of new restriction sites, specific codons, four base codons for non-natural amino acids.[5][page needed]
Transposon based random mutagenesis[edit]
Recently many methods for transposon based random mutagenesis have been reported. This methods include, but are not limited to the following: PERMUTE-random circular permutation, random protein truncation, random nucleotide triplet substitution, random domain/tag/multiple amino acid insertion, codon scanning mutagenesis, and multicodon scanning mutagenesis. These aforementioned techniques all require the design of mini-Mu transposons. Thermo scientific manufactures kits for the design of these transposons.[5][page needed]
Random mutagenesis methods altering the target DNA length[edit]
These methods involve altering gene length via insertion and deletion mutations. An example is the tandem repeat insertion (TRINS) method. This technique results in the generation of tandem repeats of random fragments of the target gene via rolling circle amplification and concurrent incorporation of these repeats into the target gene.[5][page needed]
Mutator strains[edit]
Mutator strains are bacterial cell lines which are deficient in one or more DNA repair mechanisms. An example of a mutator strand is the E. coli XL1-RED.[5][page needed] This subordinate strain of E. coli is deficient in the MutS, MutD, MutT DNA repair pathways. Use of mutator strains is useful at introducing many types of mutation; however, these strains show progressive sickness of culture because of the accumulation of mutations in the strains own genome.[5][page needed]
Focused mutagenesis[edit]
Focused mutagenic methods produce mutations at predetermined amino acid residues. These techniques require and understanding of the sequence-function relationship for the protein of interest. Understanding of this relationship allows for the identification of residues which are important in stability, stereoselectivity, and catalytic efficiency.[5][page needed] Examples of methods that produce focused mutagenesis are below.
Site saturation mutagenesis[edit]
Site saturation mutagenesis is a PCR based method used to target amino acids with significant roles in protein function. The two most common techniques for performing this are whole plasmid single PCR, and overlap extension PCR.
Whole plasmid single PCR is also referred to as site directed mutagenesis (SDM). SDM products are subjected to Dpn endonuclease digestion. This digestion results in cleavage of only the parental strand, because the parental strand contains a GmATC which is methylated at N6 of adenine. SDM does not work well for large plasmids of over ten kilobases. Also, this method is only capable of replacing two nucleotides at a time.[5][page needed]
Overlap extension PCR requires the use of two pairs of primers. One primer in each set contains a mutation. A first round of PCR using these primer sets is performed and two double stranded DNA duplexes are formed. A second round of PCR is then performed in which these duplexes are denatured and annealed with the primer sets again to produce heteroduplexes, in which each strand has a mutation. Any gaps in these newly formed heteroduplexes are filled with DNA polymerases and further amplified.[5][page needed]
Sequence saturation mutagenesis (SeSaM)[edit]
Sequence saturation mutagenesis results in the randomization of the target sequence at every nucleotide position. This method begins with the generation of variable length DNA fragments tailed with universal bases via the use of template transferases at the 3′ termini. Next, these fragments are extended to full length using a single stranded template. The universal bases are replaced with a random standard base, causing mutations. There are several modified versions of this method such as SeSAM-Tv-II, SeSAM-Tv+, and SeSAM-III.[5][page needed]
Single primer reactions in parallel (SPRINP)[edit]
This site saturation mutagenesis method involves two separate PCR reaction. The first of which uses only forward primers, while the second reaction uses only reverse primers. This avoids the formation of primer dimer formation.[5][page needed]
Mega primed and ligase free focused mutagenesis[edit]
This site saturation mutagenic technique begins with one mutagenic oligonucleotide and one universal flanking primer. These two reactants are used for an initial PCR cycle. Products from this first PCR cycle are used as mega primers for the next PCR.[5][page needed]
Ω-PCR[edit]
This site saturation mutagenic method is based on overlap extension PCR. It is used to introduce mutations at any site in a circular plasmid.[5][page needed]
PFunkel-ominchange-OSCARR[edit]
This method utilizes user defined site directed mutagenesis at single or multiple sites simultaneously. OSCARR is an acronym for one pot simple methodology for cassette randomization and recombination. This randomization and recombination results in randomization of desired fragments of a protein. Omnichange is a sequence independent, multisite saturation mutagenesis which can saturate up to five independent codons on a gene.
Trimer-dimer mutagenesis[edit]
This method removes redundant codons and stop codons.
Cassette mutagenesis[edit]
This is a PCR based method. Cassette mutagenesis begins with the synthesis of a DNA cassette containing the gene of interest, which is flanked on either side by restriction sites. The endonuclease which cleaves these restriction sites also cleaves sites in the target plasmid. The DNA cassette and the target plasmid are both treated with endonucleases to cleave these restriction sites and create sticky ends. Next the products from this cleavage are ligated together, resulting in the insertion of the gene into the target plasmid. An alternative form of cassette mutagenesis called combinatorial cassette mutagenesis is used to identify the functions of individual amino acid residues in the protein of interest. Recursive ensemble mutagenesis then utilizes information from previous combinatorial cassette mutagenesis. Codon cassette mutagenesis allows you to insert or replace a single codon at a particular site in double stranded DNA.[5][page needed]
Sexual methods[edit]
Sexual methods of directed evolution involve in vitro recombination which mimic natural in vivo recombination. Generally these techniques require high sequence homology between parental sequences. These techniques are often used to recombine two different parental genes, and these methods do create cross overs between these genes.[5][page needed]
In vitro homologous recombination[edit]
Homologous recombination can be categorized as either in vivo or in vitro. In vitro homologous recombination mimics natural in vivo recombination. These in vitro recombination methods require high sequence homology between parental sequences. These techniques exploit the natural diversity in parental genes by recombining them to yield chimeric genes. The resulting chimera show a blend of parental characteristics.[5][page needed]
DNA shuffling[edit]
This in vitro technique was one of the first techniques in the era of recombination. It begins with the digestion of homologous parental genes into small fragments by DNase1. These small fragments are then purified from undigested parental genes. Purified fragments are then reassembled using primer-less PCR. This PCR involves homologous fragments from different parental genes priming for each other, resulting in chimeric DNA. The chimeric DNA of parental size is then amplified using end terminal primers in regular PCR.[5][page needed]
Random priming in vitro recombination (RPR)[edit]
This in vitro homologous recombination method begins with the synthesis of many short gene fragments exhibiting point mutations using random sequence primers. These fragments are reassembled to full length parental genes using primer-less PCR. These reassembled sequences are then amplified using PCR and subjected to further selection processes. This method is advantageous relative to DNA shuffling because there is no use of DNase1, thus there is no bias for recombination next to a pyrimidine nucleotide. This method is also advantageous due to its use of synthetic random primers which are uniform in length, and lack biases. Finally this method is independent of the length of DNA template sequence, and requires a small amount of parental DNA.[5][page needed]
Truncated metagenomic gene-specific PCR[edit]
This method generates chimeric genes directly from metagenomic samples. It begins with isolation of the desired gene by functional screening from metagenomic DNA sample. Next, specific primers are designed and used to amplify the homologous genes from different environmental samples. Finally, chimeric libraries are generated to retrieve the desired functional clones by shuffling these amplified homologous genes.[5][page needed]
Staggered extension process (StEP)[edit]
This in vitro method is based on template switching to generate chimeric genes. This PCR based method begins with an initial denaturation of the template, followed by annealing of primers and a short extension time. All subsequent cycle generate annealing between the short fragments generated in previous cycles and different parts of the template. These short fragments and the templates anneal together based on sequence complementarity. This process of fragments annealing template DNA is known as template switching. These annealed fragments will then serve as primers for further extension. This method is carried out until the parental length chimeric gene sequence is obtained. Execution of this method only requires flanking primers to begin. There is also no need for Dnase1 enzyme.[5][page needed]
Random chimeragenesis on transient templates (RACHITT)[edit]
This method has been shown to generate chimeric gene libraries with an average of 14 crossovers per chimeric gene. It begins by aligning fragments from a parental top strand onto the bottom strand of a uracil containing template from a homologous gene. 5′ and 3′ overhang flaps are cleaved and gaps are filled by the exonuclease and endonuclease activities of Pfu and taq DNA polymerases. The uracil containing template is then removed from the heteroduplex by treatment with a uracil DNA glcosylase, followed by further amplification using PCR. This method is advantageous because it generates chimeras with relatively high crossover frequency. However it is somewhat limited due to the complexity and the need for generation of single stranded DNA and uracil containing single stranded template DNA.[5][page needed]
Synthetic shuffling[edit]
Shuffling of synthetic degenerate oligonucleotides adds flexibility to shuffling methods, since oligonucleotides containing optimal codons and beneficial mutations can be included.[5][page needed]
In vivo Homologous Recombination[edit]
Cloning performed in yeast involves PCR dependent reassembly of fragmented expression vectors. These reassembled vectors are then introduced to, and cloned in yeast. Using yeast to clone the vector avoids toxicity and counter-selection that would be introduced by ligation and propagation in E. coli.[5][page needed]
Mutagenic organized recombination process by homologous in vivo grouping (MORPHING)[edit]
This method introduces mutations into specific regions of genes while leaving other parts intact by utilizing the high frequency of homologous recombination in yeast.[5][page needed]
Phage-assisted continuous evolution (PACE)[edit]
This method utilizes a bacteriophage with a modified life cycle to transfer evolving genes from host to host. The phage’s life cycle is designed in such a way that the transfer is correlated with the activity of interest from the enzyme. This method is advantageous because it requires minimal human intervention for the continuous evolution of the gene.[5]
In vitro non-homologous recombination methods[edit]
These methods are based upon the fact that proteins can exhibit similar structural identity while lacking sequence homology.
Exon shuffling[edit]
Exon shuffling is the combination of exons from different proteins by recombination events occurring at introns. Orthologous exon shuffling involves combining exons from orthologous genes from different species. Orthologous domain shuffling involves shuffling of entire protein domains from orthologous genes from different species. Paralogous exon shuffling involves shuffling of exon from different genes from the same species. Paralogous domain shuffling involves shuffling of entire protein domains from paralogous proteins from the same species. Functional homolog shuffling involves shuffling of non-homologous domains which are functional related. All of these processes being with amplification of the desired exons from different genes using chimeric synthetic oligonucleotides. This amplification products are then reassembled into full length genes using primer-less PCR. During these PCR cycles the fragments act as templates and primers. This results in chimeric full length genes, which are then subjected to screening.[5][page needed]
Incremental truncation for the creation of hybrid enzymes (ITCHY)[edit]
Fragments of parental genes are created using controlled digestion by exonuclease III. These fragments are blunted using endonuclease, and are ligated to produce hybrid genes. THIOITCHY is a modified ITCHY technique which utilized nucleotide triphosphate analogs such as α-phosphothioate dNTPs. Incorporation of these nucleotides blocks digestion by exonuclease III. This inhibition of digestion by exonuclease III is called spiking. Spiking can be accomplished by first truncating genes with exonuclease to create fragments with short single stranded overhangs. These fragments then serve as templates for amplification by DNA polymerase in the presence of small amounts of phosphothioate dNTPs. These resulting fragments are then ligated together to form full length genes. Alternatively the intact parental genes can be amplified by PCR in the presence of normal dNTPs and phosphothioate dNTPs. These full length amplification products are then subjected to digestion by an exonuclease. Digestion will continue until the exonuclease encounters an α-pdNTP, resulting in fragments of different length. These fragments are then ligated together to generate chimeric genes.[5][page needed]
SCRATCHY[edit]
This method generates libraries of hybrid genes inhibiting multiple crossovers by combining DNA shuffling and ITCHY. This method begins with the construction of two independent ITCHY libraries. The first with gene A on the N-terminus. And the other having gene B on the N-terminus. These hybrid gene fragments are separated using either restriction enzyme digestion or PCR with terminus primers via agarose gel electrophoresis. These isolated fragments are then mixed together and further digested using DNase1. Digested fragments are then reassembled by primerless PCR with template switching.[5][page needed]
Recombined extension on truncated templates (RETT)[edit]
This method generates libraries of hybrid genes by template switching of uni-directionally growing polynucleotides in the presence of single stranded DNA fragments as templates for chimeras. This method begins with the preparation of single stranded DNA fragments by reverse transcription from target mRNA. Gene specific primers are then annealed to the single stranded DNA. These genes are then extended during a PCR cycle. This cycle is followed by template switching and annealing of the short fragments obtained from the earlier primer extension to other single stranded DNA fragments. This process is repeated until full length single stranded DNA is obtained.[5][page needed]
Sequence homology-independent protein recombination (SHIPREC)[edit]
This method generates recombination between genes with little to no sequence homology. These chimeras are fused via a linker sequence containing several restriction sites. This construct is then digested using DNase1. Fragments are made are made blunt ended using S1 nuclease. These blunt end fragments are put together into a circular sequence by ligation. This circular construct is then linearized using restriction enzymes for which the restriction sites are present in the linker region. This results in a library of chimeric genes in which contribution of genes to 5′ and 3′ end will be reversed as compared to the starting construct.[5][page needed]
Sequence independent site directed chimeragenesis (SISDC)[edit]
This method results in a library of genes with multiple crossovers from several parental genes. This method does not require sequence identity among the parental genes. This does require one or two conserved amino acids at every crossover position. It begins with alignment of parental sequences and identification of consensus regions which serve as crossover sites. This is followed by the incorporation of specific tags containing restriction sites followed by the removal of the tags by digestion with Bac1, resulting in genes with cohesive ends. These gene fragments are mixed and ligated in an appropriate order to form chimeric libraries.[5][page needed]
Degenerate homo-duplex recombination (DHR)[edit]
This method begins with alignment of homologous genes, followed by identification of regions of polymorphism. Next the top strand of the gene is divided into small degenerate oligonucleotides. The bottom strand is also digested into oligonucleotides to serve as scaffolds. These fragments are combined in solution are top strand oligonucleotides are assembled onto bottom strand oligonucleotides. Gaps between these fragments are filled with polymerase and ligated.[5][page needed]
Random multi-recombinant PCR (RM-PCR)[edit]
This method involves the shuffling of plural DNA fragments without homology, in a single PCR. This results in the reconstruction of complete proteins by assembly of modules encoding different structural units.[5][page needed]
User friendly DNA recombination (USERec)[edit]
This method begins with the amplification of gene fragments which need to be recombined, using uracil dNTPs. This amplification solution also contains primers, PfuTurbo, and Cx Hotstart DNA polymerase. Amplified products are next incubated with USER enzyme. This enzyme catalyzes the removal of uracil residues from DNA creating single base pair gaps. The USER enzyme treated fragments are mixed and ligated using T4 DNA ligase and subjected to Dpn1 digestion to remove the template DNA. These resulting dingle stranded fragments are subjected to amplification using PCR, and are transformed into E. coli.[5][page needed]
Golden Gate shuffling (GGS) recombination[edit]
This method allows you to recombine at least 9 different fragments in an acceptor vector by using type 2 restriction enzyme which cuts outside of the restriction sites. It begins with sub cloning of fragments in separate vectors to create Bsa1 flanking sequences on both sides. These vectors are then cleaved using type II restriction enzyme Bsa1, which generates four nucleotide single strand overhangs. Fragments with complementary overhangs are hybridized and ligated using T4 DNA ligase. Finally these constructs are then transformed into E. coli cells, which are screened for expression levels.[5][page needed]
Phosphoro thioate-based DNA recombination method (PRTec)[edit]
This method can be used to recombine structural elements or entire protein domains. This method is based on phosphorothioate chemistry which allows the specific cleavage of phosphorothiodiester bonds. The first step in the process begins with amplification of fragments that need to be recombined along with the vector backbone. This amplification is accomplished using primers with phosphorothiolated nucleotides at 5′ ends. Amplified PCR products are cleaved in an ethanol-iodine solution at high temperatures. Next these fragments are hybridized at room temperature and transformed into E. coli which repair any nicks.[5][page needed]
Integron[edit]
This system is based upon a natural site specific recombination system in E. coli. This system is called the integron system, and produces natural gene shuffling. This method was used to construct and optimize a functional tryptophan biosynthetic operon in trp-deficient E. coli by delivering individual recombination cassettes or trpA-E genes along with regulatory elements with the integron system.[5][page needed]
Y-Ligation based shuffling (YLBS)[edit]
This method generates single stranded DNA strands, which encompass a single block sequence either at the 5′ or 3′ end, complementary sequences in a stem loop region, and a D branch region serving as a primer binding site for PCR. Equivalent amounts of both 5′ and 3′ half strands are mixed and formed a hybrid due to the complementarity in the stem region. Hybrids with free phosphorylated 5′ end in 3′ half strands are then ligated with free 3′ ends in 5′ half strands using T4 DNA ligase in the presence of 0.1 mM ATP. Ligated products are then amplified by two types of PCR to generate pre 5′ half and pre 3′ half PCR products. These PCR product are converted to single strands via avidin-biotin binding to the 5′ end of the primes containing stem sequences that were biotin labeled. Next, biotinylated 5′ half strands and non-biotinylated 3′ half strands are used as 5′ and 3′ half strands for the next Y-ligation cycle.[5][page needed]
Semi-rational design[edit]
Semi-rational design uses information about a proteins sequence, structure and function, in tandem with predictive algorithms. Together these are used to identify target amino acid residues which are most likely to influence protein function. Mutations of these key amino acid residues create libraries of mutant proteins that are more likely to have enhanced properties.[11]
Advances in semi-rational enzyme engineering and de novo enzyme design provide researchers with powerful and effective new strategies to manipulate biocatalysts. Integration of sequence and structure based approaches in library design has proven to be a great guide for enzyme redesign. Generally, current computational de novo and redesign methods do not compare to evolved variants in catalytic performance. Although experimental optimization may be produced using directed evolution, further improvements in the accuracy of structure predictions and greater catalytic ability will be achieved with improvements in design algorithms. Further functional enhancements may be included in future simulations by integrating protein dynamics.[11]
Biochemical and biophysical studies, along with fine-tuning of predictive frameworks will be useful to experimentally evaluate the functional significance of individual design features. Better understanding of these functional contributions will then give feedback for the improvement of future designs.[11]
Directed evolution will likely not be replaced as the method of choice for protein engineering, although computational protein design has fundamentally changed the way protein engineering can manipulate bio-macromolecules. Smaller, more focused and functionally-rich libraries may be generated by using in methods which incorporate predictive frameworks for hypothesis-driven protein engineering. New design strategies and technical advances have begun a departure from traditional protocols, such as directed evolution, which represents the most effective strategy for identifying top-performing candidates in focused libraries. Whole-gene library synthesis is replacing shuffling and mutagenesis protocols for library preparation. Also highly specific low throughput screening assays are increasingly applied in place of monumental screening and selection efforts of millions of candidates. Together, these developments are poised to take protein engineering beyond directed evolution and towards practical, more efficient strategies for tailoring biocatalysts.[11]
Screening and selection techniques[edit]
Once a protein has undergone directed evolution, ration design or semi-ration design, the libraries of mutant proteins must be screened to determine which mutants show enhanced properties. Phage display methods are one option for screening proteins. This method involves the fusion of genes encoding the variant polypeptides with phage coat protein genes. Protein variants expressed on phage surfaces are selected by binding with immobilized targets in vitro. Phages with selected protein variants are then amplified in bacteria, followed by the identification of positive clones by enzyme linked immunosorbent assay. These selected phages are then subjected to DNA sequencing.[5][page needed]
Cell surface display systems can also be utilized to screen mutant polypeptide libraries. The library mutant genes are incorporated into expression vectors which are then transformed into appropriate host cells. These host cells are subjected to further high throughput screening methods to identify the cells with desired phenotypes.[5][page needed]
Cell free display systems have been developed to exploit in vitro protein translation or cell free translation. These methods include mRNA display, ribosome display, covalent and non covalent DNA display, and in vitro compartmentalization.[5]: 53
Enzyme engineering[edit]
Enzyme engineering is the application of modifying an enzyme’s structure (and, thus, its function) or modifying the catalytic activity of isolated enzymes to produce new metabolites, to allow new (catalyzed) pathways for reactions to occur,[12] or to convert from some certain compounds into others (biotransformation). These products are useful as chemicals, pharmaceuticals, fuel, food, or agricultural additives.
An enzyme reactor [13] consists of a vessel containing a reactional medium that is used to perform a desired conversion by enzymatic means. Enzymes used in this process are free in the solution. Also Microorganisms are one of important origin for genuine enzymes .[14]
Examples of engineered proteins[edit]
Computing methods have been used to design a protein with a novel fold, named Top7,[15] and sensors for unnatural molecules.[16] The engineering of fusion proteins has yielded rilonacept, a pharmaceutical that has secured Food and Drug Administration (FDA) approval for treating cryopyrin-associated periodic syndrome.
Another computing method, IPRO, successfully engineered the switching of cofactor specificity of Candida boidinii xylose reductase.[17] Iterative Protein Redesign and Optimization (IPRO) redesigns proteins to increase or give specificity to native or novel substrates and cofactors. This is done by repeatedly randomly perturbing the structure of the proteins around specified design positions, identifying the lowest energy combination of rotamers, and determining whether the new design has a lower binding energy than prior ones.[18]
Computation-aided design has also been used to engineer complex properties of a highly ordered nano-protein assembly.[19] A protein cage, E. coli bacterioferritin (EcBfr), which naturally shows structural instability and an incomplete self-assembly behavior by populating two oligomerization states, is the model protein in this study. Through computational analysis and comparison to its homologs, it has been found that this protein has a smaller-than-average dimeric interface on its two-fold symmetry axis due mainly to the existence of an interfacial water pocket centered on two water-bridged asparagine residues. To investigate the possibility of engineering EcBfr for modified structural stability, a semi-empirical computational method is used to virtually explore the energy differences of the 480 possible mutants at the dimeric interface relative to the wild type EcBfr. This computational study also converges on the water-bridged asparagines. Replacing these two asparagines with hydrophobic amino acids results in proteins that fold into alpha-helical monomers and assemble into cages as evidenced by circular dichroism and transmission electron microscopy. Both thermal and chemical denaturation confirm that, all redesigned proteins, in agreement with the calculations, possess increased stability. One of the three mutations shifts the population in favor of the higher order oligomerization state in solution as shown by both size exclusion chromatography and native gel electrophoresis.[19]
A in silico method, PoreDesigner,[20] was successfully developed to redesign bacterial channel protein (OmpF) to reduce its 1 nm pore size to any desired sub-nm dimension. Transport experiments on the narrowest designed pores revealed complete salt rejection when assembled in biomimetic block-polymer matrices.
See also[edit]
- Display:
- Bacterial display
- Phage display
- mRNA display
- Ribosome display
- Yeast display
- Biomolecular engineering
- Enzymology
- Expanded genetic code
- Fast parallel proteolysis (FASTpp)
- Gene synthesis
- Genetic engineering
- Nucleic acid analogues
- Protein structure prediction software
- Proteomics
- Proteome
- SCOPE (protein engineering)
- Structural biology
- Synthetic biology
References[edit]
- ^ «Protein engineering — Latest research and news | Nature». www.nature.com. Retrieved 2023-01-24.
- ^ Woodley, John M. (May 2022). «Integrating protein engineering into biocatalytic process scale-up». Trends in Chemistry. 4 (5): 371–373. doi:10.1016/j.trechm.2022.02.007.
- ^ «Speeding Up the Protein Assembly Line». Genetic Engineering and Biotechnology News. 13 February 2015.
- ^ Farmer, Tylar Seiya; Bohse, Patrick; Kerr, Dianne (2017). «Rational Design Protein Engineering Through Crowdsourcing». Journal of Student Research. 6 (2): 31–38. doi:10.47611/jsr.v6i2.377. S2CID 57679002.
- ^ a b c d e f g h i j k l m n o p q r s t u v w x y z aa ab ac ad ae af ag ah ai aj ak al am an ao ap aq ar as at au av aw ax ay az ba bb bc bd be bf bg bh bi bj bk Poluri, Krishna Mohan; Gulati, Khushboo (2017). Protein Engineering Techniques. SpringerBriefs in Applied Sciences and Technology. Springer. doi:10.1007/978-981-10-2732-1. ISBN 978-981-10-2731-4.
- ^ Liu, Cassie J.; Cochran, Jennifer R. (2014), Cai, Weibo (ed.), «Engineering Multivalent and Multispecific Protein Therapeutics», Engineering in Translational Medicine, London: Springer, pp. 365–396, doi:10.1007/978-1-4471-4372-7_14, ISBN 978-1-4471-4372-7, retrieved 2021-12-08
- ^ Holliger, P.; Prospero, T.; Winter, G. (1993-07-15). ««Diabodies»: small bivalent and bispecific antibody fragments». Proceedings of the National Academy of Sciences. 90 (14): 6444–6448. Bibcode:1993PNAS…90.6444H. doi:10.1073/pnas.90.14.6444. ISSN 0027-8424. PMC 46948. PMID 8341653.
- ^ Brinkmann, Ulrich; Kontermann, Roland E. (2017-02-17). «The making of bispecific antibodies». mAbs. 9 (2): 182–212. doi:10.1080/19420862.2016.1268307. ISSN 1942-0862. PMC 5297537. PMID 28071970.
- ^ Jäckel, Christian; Kast, Peter; Hilvert, Donald (June 2008). «Protein Design by Directed Evolution». Annual Review of Biophysics. 37 (1): 153–173. doi:10.1146/annurev.biophys.37.032807.125832. PMID 18573077.
- ^ Shivange, Amol V; Marienhagen, Jan; Mundhada, Hemanshu; Schenk, Alexander; Schwaneberg, Ulrich (2009). «Advances in generating functional diversity for directed protein evolution». Current Opinion in Chemical Biology. 13 (1): 19–25. doi:10.1016/j.cbpa.2009.01.019. PMID 19261539.
- ^ a b c d Lutz, Stefan (December 2010). «Beyond directed evolution—semi-rational protein engineering and design». Current Opinion in Biotechnology. 21 (6): 734–743. doi:10.1016/j.copbio.2010.08.011. PMC 2982887. PMID 20869867.
- ^ «‘Designer Enzymes’ Created By Chemists Have Defense And Medical Uses». ScienceDaily. March 20, 2008.
- ^ [Enzyme reactors at «Enzyme reactors». Archived from the original on 2012-05-02. Retrieved 2013-11-02.] Accessed 22 May 2009.
- ^ Sharma, Anshula; Gupta, Gaganjot; Ahmad, Tawseef; Mansoor, Sheikh; Kaur, Baljinder (2021-02-17). «Enzyme Engineering: Current Trends and Future Perspectives». Food Reviews International. 37 (2): 121–154. doi:10.1080/87559129.2019.1695835. ISSN 8755-9129.
- ^ Kuhlman, Brian; Dantas, Gautam; Ireton, Gregory C.; Varani, Gabriele; Stoddard, Barry L. & Baker, David (2003), «Design of a Novel Globular Protein Fold with Atomic-Level Accuracy», Science, 302 (5649): 1364–1368, Bibcode:2003Sci…302.1364K, doi:10.1126/science.1089427, PMID 14631033, S2CID 1939390
- ^ Looger, Loren L.; Dwyer, Mary A.; Smith, James J. & Hellinga, Homme W. (2003), «Computational design of receptor and sensor proteins with novel functions», Nature, 423 (6936): 185–190, Bibcode:2003Natur.423..185L, doi:10.1038/nature01556, PMID 12736688, S2CID 4387641
- ^ Khoury, GA; Fazelinia, H; Chin, JW; Pantazes, RJ; Cirino, PC; Maranas, CD (October 2009), «Computational design of Candida boidinii xylose reductase for altered cofactor specificity», Protein Science, 18 (10): 2125–38, doi:10.1002/pro.227, PMC 2786976, PMID 19693930
- ^ The iterative nature of this process allows IPRO to make additive mutations to a protein sequence that collectively improve the specificity toward desired substrates and/or cofactors. Details on how to download the software, implemented in Python, and experimental testing of predictions are outlined in this paper: Khoury, GA; Fazelinia, H; Chin, JW; Pantazes, RJ; Cirino, PC; Maranas, CD (October 2009), «Computational design of Candida boidinii xylose reductase for altered cofactor specificity», Protein Science, 18 (10): 2125–38, doi:10.1002/pro.227, PMC 2786976, PMID 19693930
- ^ a b Ardejani, MS; Li, NX; Orner, BP (April 2011), «Stabilization of a Protein Nanocage through the Plugging of a Protein–Protein Interfacial Water Pocket», Biochemistry, 50 (19): 4029–4037, doi:10.1021/bi200207w, PMID 21488690
- ^ Chowdhury, Ratul; Ren, Tingwei; Shankla, Manish; Decker, Karl; Grisewood, Matthew; Prabhakar, Jeevan; Baker, Carol; Golbeck, John H.; Aksimentiev, Aleksei; Kumar, Manish; Maranas, Costas D. (10 September 2018). «PoreDesigner for tuning solute selectivity in a robust and highly permeable outer membrane pore». Nature Communications. 9 (1): 3661. Bibcode:2018NatCo…9.3661C. doi:10.1038/s41467-018-06097-1. PMC 6131167. PMID 30202038.
External links[edit]
- servers for protein engineering and related topics based on the WHAT IF software
- Enzymes Built from Scratch — Researchers engineer never-before-seen catalysts using a new computational technique, Technology Review, March 10, 2008