Химические методы анализа
кислот как H3BO3 (pKa = 9,2) или NH4+ (pKa = 9,24) в водных растворах с удовлетворительной погрешностью невозможно и поэтому для их определения используют специальные приёмы.
Температура и ионная сила влияют на величину скачка титрования менее заметно, чем концентрация или сила титруемой кислоты или основания. При повышении температуры константа автопротолиза воды увеличивается, поэтому величина скачка кислотно-основного титрования в водном растворе уменьшается. Аналогичным образом влияет на величину скачка титрования ионная сила раствора.
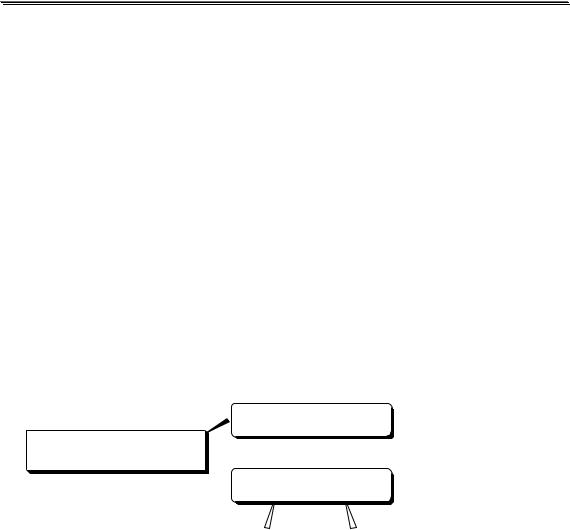
Методика титриметрического анализа многостадийна, погрешности могут возникать на любой стадии её проведения: при измерении массы навески, объёма приготовленного раствора или аликвоты, при проведении титрования, обнаружении конечной точки титрования. В зависимости от причины возникновения погрешности в титриметрических методах анализа, как и погрешности вообще, могут быть:
случайные
ПОГРЕШНОСТИ
 систематические
систематические
|
положительные |
отрицательные |
||||
|
определяемое вещество |
определяемое вещество |
||||
|
всё время перетитровывается |
всё время недотитровывается |
К появлению систематических погрешностей в титриметрических методах анализа может приводить:
•использование неверно градуированной посуды;
•неправильная техника титрования (слишком быстрое добав-
ление титранта);
•неточное считывание объёма титранта, израсходованного для титрования;
 несовпадение точки эквивалентности и рТ индикатора.
несовпадение точки эквивалентности и рТ индикатора.
Погрешности, обусловленные несовпадением точки эквивалентности и рТ индикатора, называются индикаторными.
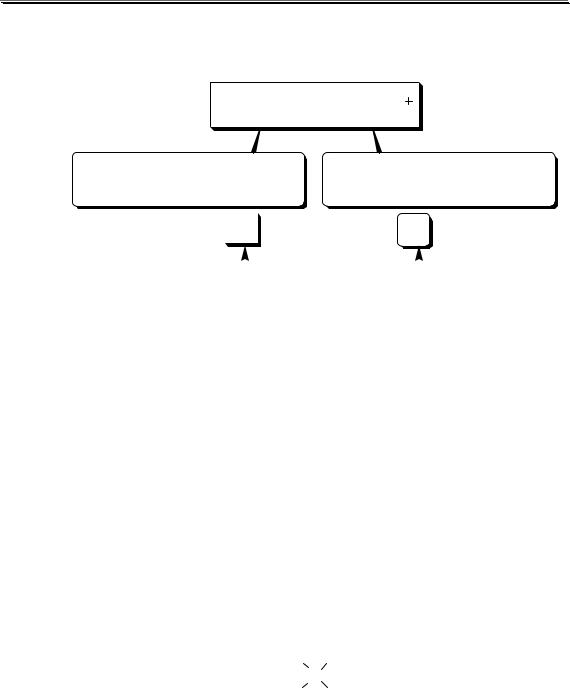
Индикаторные погрешности в кислотно-основном титровании удобно разделить на 4 вида:
161

|
Раздел 2 |
|||||||||||
|
в КТТ в растворе присутствует |
в КТТ в растворе остаётся |
||||||||||
|
избыток OH— по сравнению с ТЭ |
недотитрованная слабая кислота |
||||||||||
|
гидроксидная |
кислотная |
||||||||||
|
ИНДИКАТОРНАЯ |
|||||||||||
|
водородная |
ПОГРЕШНОСТЬ |
основная |
|||||||||
|
в КТТ в растворе присутствует |
в КТТ в растворе остаётся |
||||||||||
|
избыток H3O |
+ |
по сравнению с ТЭ |
недотитрованное слабое основание |
||||||||
Водородная индикаторная погрешность может возникнуть при недотитровании сильной кислоты (в таком случае погрешность отрицательная) либо когда сильная кислота используется в качестве титранта и добавлена в избытке (положительная погрешность).
|
n(H |
3 |
O+ ) |
конечн |
C |
V |
10 |
−pT V |
||||
|
∆ |
H |
+ = ± |
= ± |
2 конечн |
= ± |
конечн |
|||||
|
С0V0 |
|||||||||||
|
n(H3O+ )исх |
С0V0 |
Если концентрации титруемого вещества и титранта одинаковы, то Vконечн = 2V0, тогда
|
2 10−pT |
||||
|
∆ |
H |
+ = ± |
[ 100%] |
|
|
С0 |
||||
Гидроксидная погрешность может возникнуть при недотитровании сильного основания (отрицательная погрешность) либо в том случае, когда сильное основание используется в качестве титранта и добавлено в избытке (положительная погрешность).
|
10pT−pKW V |
10pT−pKW |
||||||
|
∆ |
OH |
− = ± |
конечн |
≈ ±2 |
[ 100%] |
||
|
C0 V0 |
C0 |
||||||
Кислотная и основная индикаторные погрешности могут быть только отрицательными (если, конечно, исключить гипотетический случай использования слабой кислоты или основания в качестве титранта).
Величина кислотной погрешности представляет собой молярную долю неоттитрованной кислоты.
|
[HA] |
[H |
3O+ ] |
||||||||
|
∆HA = − |
= − |
= |
− |
1 |
[ 100%] |
|||||
|
pT−pKa |
||||||||||
|
C(HA) |
+ |
+10 |
||||||||
|
[H3O |
] + K a |
1 |
||||||||
162

Химические методы анализа
Если 10pT−pKa >>1, то ∆HA = −10pKa −pH [ 100%]
Формула для расчёта основной погрешности выводится аналогичным образом и выглядит следующим образом
|
∆B = − |
1 |
[ 100%] |
||||
|
1 +10 |
pK |
BH |
+ −pT |
|||
или в упрощённом виде
∆B = −10pT−pKBH+ [100%]
Пример 13.1. Рассчитать систематическую индикаторную погрешность титрования 0,1 М HCl и 0,1 М HCOOH при использовании в качестве титранта 0,1 М NaOH и индикатора метилового оранжевого (рТ = 4).
В случае HCl титрование заканчивается при рН меньшем (4), чем рН в точке эквивалентности (7), поэтому имеет место водородная индикаторная погрешность. Поскольку в конечной точке титрования определяемое вещество будет недотитровано, величина систематической индикаторной погрешности будет отрицательной
∆H+ = −2 1 10−4 100% = −0,2% 0,1
При титровании HCOOH в конечной точке титрования будет оставаться неоттитрованная слабая кислота, поэтому в данном случае будет кислотная индикаторная погрешность.
|
∆HA = − |
1 |
100% |
= −36% |
|
|
+100,25 |
||||
|
1 |
Совершенно очевидно, что метиловый оранжевый не может быть использован для обнаружения конечной точки титрования раствора HCOOH раствором NaOH.
Даже в том случае, если систематическая индикаторная погрешность равна 0 (pHэкв = рТ), всё равно будет иметься случайная погрешность визуального обнаружения конечной точки титрования с помощью индикатора. Вследствие физиологических особенностей нашего зрения рТ индикатора можно определить лишь с неопределённостью примерно ± 0,4 ед. рН. Величина случайной индикаторной погрешности зависит от крутизны скачка титрования — чем она больше, тем случайная погрешность меньше. Индекс крутизны скачка титрования рассчитывается следующим образом:
163

Раздел 2
η = dpHdf ≈ ∆∆рНf
При титровании слабых кислот (оснований) крутизна скачка титрования меньше, следовательно, случайная индикаторная погрешность больше, чем при титровании сильных кислот (оснований) (рис 13.5). Для 0,1 М сильных кислот и оснований величина случайной индикаторной погрешности составляет ±2 10-7. По мере уменьшения силы кислоты (основания) и концентрации случайная погрешность увеличивается.
|
pH |
|||
|
10 |
|||
|
9 |
|||
|
8 |
|||
|
7 |
1 |
||
|
6 |
|||
|
5 |
|||
|
4 |
2 |
||
|
3 |
f |
||
|
0,99 |
1 |
1,01 |
Рис. 13.5. Влияние крутизны скачка титрования на случайную индикатор-
ную погрешность: 1 – 0,1 М HCOOH; 2 – 0,1 М HCl
В виде полосы показана область неопределённости обнаружения конечной точки титрования для индикатора, имеющего рТ 8
13.6. Некоторые случаи практического применения кислотно-основного титрования в водных растворах
Анализ смеси карбоната и гидроксида, карбоната и гидрокарбоната щелочного металла с применением двух индикаторов
При титровании смеси гидроксида и карбоната щелочного металла, например, NaOH и Na2CO3 и обнаружении конечной точки титрования с помощью фенолфталеина протекают реакции:
NaOH + HCl → NaCl + H2O
Na2CO3 + HCl → NaHCO3 + NaCl,
При обнаружении конечной точки титрования с помощью метилового оранжевого реакция взаимодействия гидроксида натрия с кислотой протекает точно также, а карбонат натрия титруется до уголь-
164

Химические методы анализа
ной кислоты. Разность между объёмами раствора титранта, израсходованного для титрования смеси в присутствии метилового оранжевого и фенолфталеина, будет соответствовать протеканию реакции:
NaHCO3 + HCl → H2CO3 + NaCl
Фактор эквивалентности NaHCO3 в данной реакции равен 1. Если принять, что NaHCO3 в исходной смеси не было, то n(NaHCO3) = n0(Na2CO3) и массу карбоната натрия можно рассчитать следующим образом
m(Na2CO3) = C(HCl) (VМО – VФ) 10-3 M(Na2CO3)
Для взаимодействия с NaOH, находящимся в анализируемой пробе, будет расходоваться объём стандартного раствора титранта
равный VФ – (VМО – VФ) = 2VФ — VМО, поэтому массу NaOH рассчитывают по следующей формуле
m(NaOH) = C(HCl) (2VФ – VМО) 10-3 M(NaOH)
Если на титрование смеси щелочи и карбоната с фенолфталеином и метиловым оранжевым затрачивается практически одинаковый объём стандартного раствора титранта, то содержание карбоната в смеси очень мало. Напротив, если объёмы раствора титранта, затраченные для титрования, значительно отличаются, то в анализируемой смеси содержится много карбоната
Анализ смеси гидрокарбоната и карбоната щелочного металла титрованием её раствором сильной кислоты в присутствии двух индикаторов основан на том же принципе, что и анализ смеси гидроксида и карбоната. При титровании смеси с фенолфталеином с титрантом взаимодействует лишь карбонат
Na2CO3 + HCl → NaHCO3 + NaCl
С метиловым оранжевым титруются и карбонат и гидрокарбонат. По объёму раствора HCl, затраченному для титрования с фенолфталеином, можно рассчитать содержание Na2CO3 (fэкв = 1), а по разности между объёмом раствора HCl, затраченным для титрования с метиловым оранжевым и удвоенным объёмом, затраченным для титрования с фенолфталеином — содержание NaHCO3:
m(Na2CO3) = C(HCl) VФ 10-3 M(Na2CO3) m(NaHCO3) = C(HCl) (VМО – 2VФ) 10-3 M(NaHCO3)
Чем больше титранта требуется для титрования с фенолфталеином, тем больше карбоната содержится в анализируемой пробе. Если при добавлении к титруемому раствору фенолфталеина последний окрашивается в слабо розовый цвет и для его обесцвечивания требуется лишь несколько капель раствора титранта, то содержание карбоната в пробе очень мало.
165

Раздел 2
Определение азота в органических соединениях по Кьельдалю и ионов аммония
Определение азота в органических соединениях методом Кьельдаля проводят следующим образом (устройство прибора показано на рис. 13.6). Точную навеску анализируемого образца помещают в колбу Кьедьдаля и подвергают минерализации с помощью концентрированной серной кислоты, к которой добавлены K2SO4 и СuSO4, а в некоторых случаях ещё и селен или HgO. В процессе окисления органической части молекулы азот восстанавливается до иона аммония. По-
|
сле окончания минерализации к раство- |
||||
|
ру добавляют NaOH. При этом образу- |
||||
|
ется NH3, который отгоняют и погло- |
||||
|
щают раствором H3BO3 или стандарт- |
||||
|
ным раствором сильной кислоты (H2SO4 |
||||
|
или HCl). В первом случае при взаимо- |
||||
|
действии борной кислоты с аммиаком |
||||
|
образуется эквивалентное NH3 количе- |
||||
|
ство иона BO2—, который затем титруют |
||||
|
стандартным раствором HCl (титрова- |
||||
|
ние заместителя). Во втором случае оп- |
||||
|
ределяют избыток сильной кислоты, не |
||||
|
вступивший в реакцию с NH3, титруя |
||||
|
Рис. 13.6. Прибор для определе- |
раствор стандартным раствором NaOH |
|||
|
ния азота в органических соеди- |
(обратное титрование). |
|||
|
нениях (по ГФ XI) |
Обычный метод Кьельдаля ис- |
|||
|
1 – парообразователь; 2 – колба |
пользуют для органических соединений, |
|||
|
Кьельдаля; 3 – воронка для вво- |
содержащих аминный азот (амины, |
|||
|
да щелочи; 4 – брызгоуловитель; |
аминокислоты и т.д.). Для определения |
|||
|
5 – холодильник; 6 — приёмник |
||||
|
азота в нитратах, нитритах, нитросоеди- |
нениях и т.п. необходимо ещё предварительное восстановление данных азотсодержащих групп до иона аммония или аминогруппы.
Методику, похожую на описанную выше, можно использовать также и для веществ, которые легко гидролизуются с образованием аммиака или аминов. Такие вещества не подвергают минерализации, а сразу проводят их щелочной гидролиз. Например, определение азота в соединении (1) требует обязательной минерализации, а для соединения (2) достаточно щелочного гидролиза.
|
O |
O |
||
|
C |
NH |
C |
NH2 |
|
(1) |
(2) |
||
|
OH |
OH |
166
Химические методы анализа
Ион аммония является достаточно слабой кислотой (pKa = 9,24), поэтому его прямое титриметрическое определение при концентрации в водном растворе, например, 0,1 моль/л, невозможно.
ОПРЕДЕЛЕНИЕ NH4
|
формальдегидный способ |
обратное титрование |
||||||||||
|
(титрование заместителя) |
|||||||||||
|
+ |
∆ |
||||||||||
|
+ 6CH2O → (CH2)6N4 + |
+ |
NH |
+ |
+ OH |
— |
↑ + H O |
|||||
|
4NH4 |
4H + 6H2O |
4 |
→ NH |
||||||||
|
3 |
2 |
||||||||||
|
титруют стандартным раствором |
избыток щелочи, не вступивший в |
||||||||||
|
щёлочи в присутствии фенолфталеина |
реакцию, титруют стандартным |
||||||||||
|
раствором сильной кислоты |
Определение борной кислоты
Борная кислота является слабой одноосновной кислотой (pKa ≈ 9,3). Её кислотные свойства обусловлены реакцией:
B(OH)3 + 2H2O î [B(OH)4]— + H3O+
Борная кислота является слишком слабой для того, чтобы её можно было с удовлетворительной погрешностью оттитровать щелочью в водном растворе. Однако, она может взаимодействовать с органическими веществами, в состав которых входит α-диольная группа (глицерин, глюкоза, фруктоза, маннит, сорбит и др.), с образованием более сильных комплексных кислот (например, у маннитборной кислоты pKa = 5,3). Последние могут быть оттитрованы раствором щёлочи в присутствии фенолфталеина.
|
C |
OH |
C |
O |
O |
C |
+ H |
O+ + 2H O |
|||||||||||||||||
|
2 |
+ B(OH) |
î |
B |
|||||||||||||||||||||
|
C |
OH |
3 |
3 |
2 |
||||||||||||||||||||
|
C |
O |
O |
C |
|||||||||||||||||||||
Наиболее часто используемым на практике комплексообразователем при определении борной кислоты является глицерин, хотя по сравнению, например, с маннитом или моносахаридами данное вещество является менее активным комплексообразующим реагентом. Кроме того, глицерин очень вязкий и работать с ним неудобно. Глицерин, используемый в лаборатории, может содержать примеси кислот. Перед применением его необходимо нейтрализовать раствором щёлочи до появления слабо-розового окрашивания фенолфталеина.
167
Соседние файлы в предмете [НЕСОРТИРОВАННОЕ]
- #
- #
- #
- #
- #
- #
- #
- #
- #
- #
- #
|
|

Макеты страниц
Индикаторная ошибка. Изменение цвета индикатора происходит не абсолютно точно в точке эквивалентности, а раньше или позже, поэтому химик-аналитик допускает некоторую ошибку, прекращая титрование раньше или позже требуемого момента. Такую ошибку называют индикаторной ошибкой. Величина ее может колебаться в самых широких пределах в зависимости от того, насколько удачно выбран примененный индикатор.
Предположим, что для титрования 0,1 н. раствора слабой кислоты с  н. раствором сильной щелочи мы использовали два индикатора: метиловый оранжевый
н. раствором сильной щелочи мы использовали два индикатора: метиловый оранжевый  и фенолфталеин
и фенолфталеин  . Посмотрим, что произойдет в обоих случаях титрования.
. Посмотрим, что произойдет в обоих случаях титрования.  исходной кислоты равен 2,87; в точке эквивалентности
исходной кислоты равен 2,87; в точке эквивалентности  (см. § 16).
(см. § 16).
В случае прибавления к титруемой кислоте метилового оранжевого раствор окрасится в красный цвет. Вскоре после прибавления первых порций сильного основания  раствора станет равным 3,1 и раствор окрасится в оранжевый цвет, а при
раствора станет равным 3,1 и раствор окрасится в оранжевый цвет, а при  раствор окрасится в желтый цвет, не изменяющийся при дальнейшем добавлении щелочи. Следовательно, после прибавления нескольких миллилитров, а быть может и капель щелочи придется прекратить титрование раньше достижения точки эквивалентности. Рассчитанное количество титруемой кислоты может оказаться в несколько раз ниже, чем действительное ее содержание, и индикаторная ошибка может достичь 75—85%.
раствор окрасится в желтый цвет, не изменяющийся при дальнейшем добавлении щелочи. Следовательно, после прибавления нескольких миллилитров, а быть может и капель щелочи придется прекратить титрование раньше достижения точки эквивалентности. Рассчитанное количество титруемой кислоты может оказаться в несколько раз ниже, чем действительное ее содержание, и индикаторная ошибка может достичь 75—85%.
Если же к титруемой кислоте прибавить фенолфталеин, то раствор будет оставаться бесцветным до тех пор, пока по мере прибавления к нему щелочи  раствора не достигнет 8,0. При
раствора не достигнет 8,0. При  раствор порозовеет, а при
раствор порозовеет, а при  раствор окрасится в красный цвет.
раствор окрасится в красный цвет.
Так как в точке эквивалентности  что укладывается в интервал
что укладывается в интервал  , то рассчитанное количество кислоты будет практически отвечать действительному ее содержанию и индикаторная ошибка составит
, то рассчитанное количество кислоты будет практически отвечать действительному ее содержанию и индикаторная ошибка составит  .
.
Таким образом, если индикатор выбран правильно, то индикаторную ошибку не принимают во внимание; если же индикатор выбран неправильно, то индикаторная ошибка превышает допустимые погрешности и может достигать очень большой величины. В случаях, требующих более точных результатов, следует учитывать индикаторные ошибки.
Типы индикаторных ошибок. К индикаторным ошибкам относят такие, которые вызываются недотитрованием или перетитрованием исследуемого раствора. В методе нейтрализации различают несколько типов индикаторных ошибок.
а) Водородная ошибка титрования, вызываемая наличием в титруемом растворе по окончании титрования избытка ионов водорода, остающихся в растворе в результате недотитрования сильной кислоты сильной щелочью (обозначается  -ошибка) или перетитрования сильного основания сильной кислотой (обозначается
-ошибка) или перетитрования сильного основания сильной кислотой (обозначается  -ошибка).
-ошибка).
б) Гидроксильная ошибка титрования, вызываемая наличием в титруемом растворе по окончании титрования избытка ионов гидроксила, остающихся в растворе в результате недотитрования сильного основания сильной кислотой ( -ошибка) или перетитрования сильной кислоты сильной щелочью (
-ошибка) или перетитрования сильной кислоты сильной щелочью ( -ошибка).
-ошибка).
в) Кислотная ошибка титрования, вызываемая присутствием в титруемом растворе по окончании титрования нейтральных молекул недотитрованной слабой кислоты ( -ошибка).
-ошибка).
г) Щелочная ошибка титрования, вызываемая присутствием в титруемом растворе по окончании титрования нейтральных молекул недотитрованного слабого основания ( -ошибка).
-ошибка).
Примеры вычисления ошибок титрования.  шибка. Предположим, что для титрования сильной кислоты сильным основанием выбран индикатор с
шибка. Предположим, что для титрования сильной кислоты сильным основанием выбран индикатор с  (например, метиловый оранжевый). Титрование заканчивается при
(например, метиловый оранжевый). Титрование заканчивается при  , т. е. при
, т. е. при  (в кислой среде). Следовательно, часть титруемой кислоты будет недотитрована и мы допустим
(в кислой среде). Следовательно, часть титруемой кислоты будет недотитрована и мы допустим  -ошибку. Вычислим ее величину.
-ошибку. Вычислим ее величину.
Пусть концентрация титруемой кислоты  начальный объем кислоты
начальный объем кислоты  объем раствора в конце титрования
объем раствора в конце титрования  .
.
Каждый миллилитр 0,1 н. раствора кислоты или щелочи содержит  г-экв. Для
г-экв. Для  титрования взято
титрования взято  г-экв кислоты.
г-экв кислоты.
Неоттитрованных ионов водорода  останется
останется  , или
, или  раствора
раствора  г-экв кислоты. Эта величина и составляет водородную ошибку титрования, обусловливаемую недотитрованием
г-экв кислоты. Эта величина и составляет водородную ошибку титрования, обусловливаемую недотитрованием  -ионов.
-ионов.
Величину  -ошибки (в процентах) вычисляют согласно пропорции:
-ошибки (в процентах) вычисляют согласно пропорции:  ошибка
ошибка 

В нашем примере

 -шибка. Предположим, что для титрования сильной кислоты сильным основанием выбран индикатор с
-шибка. Предположим, что для титрования сильной кислоты сильным основанием выбран индикатор с  (например, фенолфталеин). В этом случае титрование заканчивается при
(например, фенолфталеин). В этом случае титрование заканчивается при  , т. е. при
, т. е. при  щелочной среде. Следовательно, при титровании будет прилит некоторый избыток щелочи, что приведет к
щелочной среде. Следовательно, при титровании будет прилит некоторый избыток щелочи, что приведет к  -ошибке. Вычислим ее величину.
-ошибке. Вычислим ее величину.
Концентрация щелочи к концу титрования составит:

или

В  раствора
раствора  г-экв щелочи.
г-экв щелочи.
Эта величина и составляет гидроксильную ошибку титрования, обусловливаемую перетитрованием кислоты щелочью.
В процентах:

В нашем примере (при  :
:

Это означает, что при титровании сильной кислоты сильным основанием в присутствии метилового оранжевого изменение окраски индикатора наступает раньше, а при титровании в присутствии фенолфталеина (при условии отсутствия в растворе  ) — после достижения точки эквивалентности, причем ошибка титрования при использовании фенолфталеина в 10 раз меньше ошибки титрования с индикатором метиловым оранжевым.
) — после достижения точки эквивалентности, причем ошибка титрования при использовании фенолфталеина в 10 раз меньше ошибки титрования с индикатором метиловым оранжевым.
Так как величина концентрации титруемого раствора входит в знаменатель дроби, то ошибка титрования будет тем больше, чем менее концентрированный раствор титруют.
Поэтому, чтобы избежать больших ошибок титрования, не следует титровать слишком разбавленные растворы очень разбавленными титрантами.
Влияние области интервала перехода индикатора на величину ошибки титрования. При титровании оснований в присутствии индикатора метилового оранжевого  необходимо не только прибавить требуемое по расчету количество сильной кислоты, но и добавить некоторый ее избыток, для того чтобы окраска индикатора изменилась от желтой к оранжево-красной.
необходимо не только прибавить требуемое по расчету количество сильной кислоты, но и добавить некоторый ее избыток, для того чтобы окраска индикатора изменилась от желтой к оранжево-красной.
Если  , то этот избыток (
, то этот избыток ( ) вычисляют по формуле:
) вычисляют по формуле:

При  эта величина возрастает до
эта величина возрастает до  . Такая ошибка совершенно недопустима. Поэтому, чтобы избежать слишком больших ошибок титрования, не следует применять при титровании оснований индикаторы, интервалы перехода которых лежат ниже
. Такая ошибка совершенно недопустима. Поэтому, чтобы избежать слишком больших ошибок титрования, не следует применять при титровании оснований индикаторы, интервалы перехода которых лежат ниже  .
.
При титровании кислоты в присутствии фенолфталеина  необходимо прибавить некоторый избыток щелочи, чтобы индикатор из бесцветной формы перешел в окрашенную. Избыток
необходимо прибавить некоторый избыток щелочи, чтобы индикатор из бесцветной формы перешел в окрашенную. Избыток  находят по формуле:
находят по формуле:

При  , равном 11 и 12, эта величина возрастает до 0,5 и
, равном 11 и 12, эта величина возрастает до 0,5 и  . Такая ошибка совершенно недопустима.
. Такая ошибка совершенно недопустима.
Поэтому, чтобы избежать слишком больших ошибок титрования, не следует применять при титровании кислот индикаторы, интервалы перехода которых лежат выше  .
.
Гидроксильная ошибка титрования. Предположим, что для титрования сильного основания сильной кислотой выбран индикатор с  концентрация титруемого основания
концентрация титруемого основания  начальный объем основания
начальный объем основания  объем в конце титрования
объем в конце титрования  .
.
Каждый миллилитр 0,1 н. раствора основания содержит г-экв.
Для титрования взято  г-экв основания. Титрование заканчивают при
г-экв основания. Титрование заканчивают при  (в щелочной среде); следовательно,
(в щелочной среде); следовательно,  , т. е.
, т. е.  . Таким образом, часть титруемой щелочи будет недотитрована, и мы допустим
. Таким образом, часть титруемой щелочи будет недотитрована, и мы допустим  -ошибку. Вычислим ее величину.
-ошибку. Вычислим ее величину.
Концентрация ионов гидроксила в конце титрования составит  или
или  .
.
В  раствора
раствора  основания.
основания.
Эта величина и составляет гидроксильную ошибку титрования, обусловленную недотитрованием ОН « -ионов.
-ионов.
Величину  -ошибки (в процентах) вычисляют по формуле:
-ошибки (в процентах) вычисляют по формуле:

В нашем примере

Если выполнять титрование в присутствии индикатора с  , то необходимо прибавить некоторый избыток кислоты.
, то необходимо прибавить некоторый избыток кислоты.
Водородная ошибка титрования. Предположим, что для титрования сильного основания сильной кислотой выбран индикатор  . Титрование заканчивают при
. Титрование заканчивают при  — составляет водородную ошибку титрования, обусловленную перетитрованием щелочи кислотой.
— составляет водородную ошибку титрования, обусловленную перетитрованием щелочи кислотой.
В процентах

 — ошибка
— ошибка  (57)
(57)
Если  , то в этом случае получим:
, то в этом случае получим:

Кислотная ошибка титрования. Предположим, что для титрования дана слабая кислота  . Титрование ведут в присутствии индикатора с
. Титрование ведут в присутствии индикатора с  . В этом случае
. В этом случае

или

Так как  для слабого электролита равна
для слабого электролита равна  , то можно написать:
, то можно написать:

 Титрование заканчивается при
Титрование заканчивается при  , следовательно,
, следовательно,  -ошибка равна:
-ошибка равна:

Отношение можно рассматривать как отношение концентраций неоттитрованной части кислоты к оттитрованной ее части и считать это отношение критерием кислотной ошибки титрования. В нашем примере  -ошибка
-ошибка  или
или  . При титровании той же самой кислоты в присутствии индикатора с
. При титровании той же самой кислоты в присутствии индикатора с  соответствующая ошибка увеличилась бы в
соответствующая ошибка увеличилась бы в  :
:

Пример. Вычислите ошибку титрования 0,1 н. раствора  0,1 н. раствором
0,1 н. раствором  в присутствии индикатора метилового оранжевого
в присутствии индикатора метилового оранжевого  :
:

Следовательно, на каждую оттитрованную молекулу  приходится 5,5 неоттитрованных.
приходится 5,5 неоттитрованных.

Это значит, что 85% кислоты будет не оттитровано, если титровать  раствором
раствором  в присутствии метилового оранжевого. Следовательно, титровать
в присутствии метилового оранжевого. Следовательно, титровать  с индикаторами, имеющими
с индикаторами, имеющими  , нельзя.
, нельзя.
Щелочная ошибка титрования. Предположив, что для титрования дано слабое основание КЮН, щелочную ошибку титрования вычисляют аналогично кислотной ошибке:
 (59)
(59)
Пример. Вычислите ошибку титрования 0,1 н. раствора аммиака  0,1 н. раствором
0,1 н. раствором  в присутствии индикатора фенолфталеина
в присутствии индикатора фенолфталеина  :
:

Следовательно, на каждую оттитрованную молекулу  приходится 0,55 неоттитрованной:
приходится 0,55 неоттитрованной:

Это значит, что около 35% аммиака будет не оттитровано, если его титровать раствором  в присутствии фенолфталеина. Следовательно, титровать раствор
в присутствии фенолфталеина. Следовательно, титровать раствор  с индикатором, имеющим
с индикатором, имеющим  , нельзя.
, нельзя.
Оглавление
- ПРЕДИСЛОВИЕ К ПЕРВОМУ ИЗДАНИЮ
- ПРЕДИСЛОВИЕ КО ВТОРОМУ ИЗДАНИЮ
- ПРЕДИСЛОВИЕ К ТРЕТЬЕМУ ИЗДАНИЮ
- ВВЕДЕНИЕ
- § 1. Понятие о количественном анализе
- § 2. Классификация методов количественного анализа
- § 3. Характеристика методов количественного анализа
- § 4. Анализ больших и малых количеств вещества
- § 5. Отбор средней пробы
- § 6. Подготовка вещества для взвешивания
- § 7. Взвешивание
- § 8. Техника взвешивания на аналитических весах
- § 9. Правила обращения с аналитическими весами
- § 10. Приготовление раствора для анализа
- § 11. Запись результатов анализа
- Часть первая. Объемный анализ
- § 1. Сущность объемного анализа
- § 2. Общее уравнение реакции титрования и выводы из него
- Б. ТЕХНИКА ХИМИЧЕСКОГО ЭКСПЕРИМЕНТА В ОБЪЕМНОМ АНАЛИЗЕ
- § 3. Измерение объемов растворов
- § 4. Посуда, применяемая для измерения объемов растворов
- § 5. Работа с мерными колбами
- § 6. Работа с пипетками
- § 7. Работа с бюретками
- § 8. Приготовление стандартных растворов
- В. ВЫЧИСЛЕНИЯ В ОБЪЕМНОМ АНАЛИЗЕ
- § 9. Концентрация растворов и способы ее выражения
- § 10. Способы вычисления в объемном анализе
- § 11. Связь между точностью измерений и точностью вычислений
- § 12. Краткие сведения о статистической обработке экспериментальных данных
- Г. ПОЛУМИКРООБЪЕМНЫЙ МЕТОД АНАЛИЗА
- § 13. Понятие о полумикрообъемном анализе
- § 14. Особенности техники измерения объемов растворов в полумикрометоде
- Д. БЕЗБЮРЕТОЧНЫЕ МЕТОДЫ ТИТРОВАНИЯ
- § 15. Понятие о безбюреточных методах титрования
- § 16. Классификация методов безбюреточного титрования
- Е. АВТОМАТИЧЕСКИЕ МЕТОДЫ
- § 17. Химико-аналитический контроль производства
- § 18. Автоматические методы титрования
- ГЛАВА II. МЕТОДЫ НЕЙТРАЛИЗАЦИИ, ИЛИ МЕТОДЫ КИСЛОТНО-ОСНОВНОГО ТИТРОВАНИЯ
- § 1. Характеристика метода
- § 2. Установление точки эквивалентности
- § 3. Графический метод изображения процесса нейтрализации
- § 4. Вычисление концентрации ионов водорода в водных растворах сильных кислот и оснований
- § 5. Вычисление активности ионов водорода в водных растворах сильных кислот и оснований
- § 6. Титрование сильной кислоты сильным основанием
- § 7. Вычисление концентрации ионов водорода в растворах слабых кислот и оснований
- § 8. Вычисление активности ионов водорода в водных растворах слабых кислот и оснований
- § 9. Равновесия в водных буферных растворах слабых кислот в присутствии солей этих кислот
- § 10. Равновесия в водных буферных растворах слабых оснований в присутствии солей этих оснований
- § 11. Вычисление концентрации ионов водорода в водных буферных растворах
- § 12. Вычисление активности ионов водорода в водных буферных растворах
- § 13. Вычисление концентрации ионов водорода и степени гидролиза в водных растворах гидролизующихся бинарных солей
- § 14. Вычисление активности ионов водорода в водных растворах гидролизующихся бинарных солей
- § 15. Титрование слабой кислоты сильным основанием
- § 16. Титрование слабого основания сильной кислотой
- § 17. Титрование многоосновных кислот
- § 18. Титрование солей, образованных катионами сильных оснований и анионами слабых многоосновных кислот
- § 19. Изменение активности и показателя активности ионов водорода в процессе титрования водных растворов кислот и оснований
- § 20. Выводы, вытекающие из рассмотрения кривых нейтрализации
- § 21. Индикаторы
- § 22. Интервал перехода индикатора
- § 23. Выбор индикатора
- § 24. Ошибки титрования
- Б. ПРАКТИЧЕСКАЯ ЧАСТЬ
- § 25. Организация рабочего места
- § 26. Приготовление стандартных (титрованных) растворов
- § 27. Приготовление 0,1 н. раствора хлористоводородной кислоты
- § 28. Установка титра 0,1 н. раствора хлористоводородной кислоты
- § 29. Приготовление 0,1 н. раствора едкого натра
- § 30. Установка титра 0,1 н. раствора едкого натра
- § 31. Определение карбонатов
- § 32. Определение содержания H2SO4 в технической серной кислоте
- § 33. Определение содержания уксусной кислоты
- § 34. Определение содержания Na2CO3 и NaOH при их совместном присутствии
- § 35. Определение содержания Na2CO3 и NaHCO3 при их совместном присутствии
- § 36. Определение жесткости воды
- § 37. Определение аммонийного азота в солях аммония
- § 38. Определение содержания фосфорной кислоты
- В. КИСЛОТНО-ОСНОВНОЕ ТИТРОВАНИЕ В НЕВОДНЫХ СРЕДАХ
- § 39. Неводные растворы
- § 40. Современные представления о кислотах и основаниях
- § 41. Диссоциация электролитов в неводных растворах
- § 42. Влияние неводных растворителей на силу кислот и оснований
- § 43. Применение закона действия масс к растворам сильных электролитов
- § 44. Титрование кислот и оснований в неводных растворах
- § 45. Методы кислотно-основного титрования в неводных средах
- § 46. Примеры практических определений в неводных растворах
- ГЛАВА III. МЕТОДЫ ОКИСЛЕНИЯ—ВОССТАНОВЛЕНИЯ (ОКСИДИМЕТРИЯ, ОКСРЕДМЕТРИЯ, РЕД-ОКС-МЕТОДЫ)
- § 1. Значение окислительно-восстановительных потенциалов
- § 2. Реакции окисления—восстановления и комплексообразования
- § 3. Примеры окислительно-восстановительного титрования
- § 4. Константы равновесия окислительно-восстановительных реакций
- § 5. Связь между константами равновесия окислительно-восстановительных реакций и нормальными потенциалами
- § 6. Вычисление констант равновесия окислительно-восстановительных реакций
- § 8. Зависимость скорости реакций окисления—восстановления от различных факторов
- § 9. Графический метод изображения процесса окисления—восстановления
- § 10. Фиксирование точки эквивалентности в методах окисления—восстановления
- § 11. Окислительно-восстановительные индикаторы (ред-окс-индикаторы)
- Б. ПЕРМАНГАНАТОМЕТРИЯ
- § 12. Основы перманганатометрии
- § 13. Титрование перманганатом в кислой среде
- § 14. Титрование перманганатом в щелочной среде
- § 15. Приготовление стандартного (титрованного) раствора перманганата калия
- § 16. Установка титра стандартного раствора перманганата калия
- § 17. Установка титра и нормальности раствора перманганата калия по оксалату аммония
- § 18. Вещества, определяемые методом перманганатометрии
- ОПРЕДЕЛЕНИЕ ВОССТАНОВИТЕЛЕЙ
- § 19. Определение щавелевой кислоты и оксалатов
- § 20. Определение соединений железа (II)
- § 21. Определение содержания металлического железа в присутствии окислов железа
- § 22. Определение азотистой кислоты и нитритов
- § 23. Определение содержания марганца (II) в рудах
- ОПРЕДЕЛЕНИЕ ОКИСЛИТЕЛЕЙ
- § 24. Определение соединений железа (III)
- § 25. Определение нитратов
- § 26. Определение бихроматов
- § 27. Определение содержания MnO2 в пиролюзите
- ОПРЕДЕЛЕНИЕ ДРУГИХ ВЕЩЕСТВ
- § 28. Определение ионов кальция
- В. ИОДОМЕТРИЯ
- § 29. Основы иодометрии
- § 30. Методы иодометрического титрования
- § 31. Преимущества и недостатки иодометрического метода
- § 32. Приготовление стандартного (титрованного) раствора тиосульфата и установка его титра
- § 33. Приготовление стандартного (титрованного) раствора иода и установка его титра
- МЕТОДЫ ПРЯМОГО ТИТРОВАНИЯ
- § 34. Определение мышьяка (III)
- МЕТОДЫ ОБРАТНОГО ТИТРОВАНИЯ
- § 35. Определение сульфита натрия
- § 36. Определение содержания формальдегида в формалине
- МЕТОДЫ КОСВЕННОГО ОПРЕДЕЛЕНИЯ
- § 37. Определение ионов меди (II)
- § 38. Определение двуокиси свинца в сурике
- МЕТОД ТИТРОВАНИЯ ЗАМЕСТИТЕЛЕЙ
- § 39. Определение содержания двуокиси марганца в пиролюзите
- МЕТОД ОПРЕДЕЛЕНИЯ КИСЛОТ
- § 40. Определение хлористоводородной кислоты
- § 41. Определение воды по Фишеру
- Г. ПОНЯТИЕ О ДРУГИХ МЕТОДАХ ОКИСЛЕНИЯ — ВОССТАНОВЛЕНИЯ
- § 42. Хроматометрия
- § 43. Определение содержания железа (II)
- § 44. Цериметрия
- § 45. Броматометрия
- § 46. Ванадатометрия
- § 47. Аскорбинометрия
- § 48. Титанометрия
- ГЛАВА IV. МЕТОДЫ ОСАЖДЕНИЯ И КОМПЛЕКСООБРАЗОВАНИЯ
- § 1. Общая характеристика методов
- § 2. Классификация методов осаждения и комплексообразования
- § 3. Применение теории осаждения к объемному анализу
- § 4. Вычисление растворимости электролитов в воде с учетом коэффициентов активности
- § 5. Влияние одноименных ионов на растворимость малорастворимого электролита
- § 6. Солевой эффект
- § 7. Влияние концентрации ионов водорода на растворимость малорастворимых соединений
- § 8. Кривые титрования в методе осаждения
- § 9. Общие выводы, вытекающие из рассмотрения кривых осаждения
- § 10. Адсорбционные явления, наблюдаемые при титровании по методу осаждения
- Б. АРГЕНТОМЕТРИЯ
- § 11. Характеристика метода
- § 12. Приготовление 0,1 н. раствора нитрата серебра
- § 13. Приготовление стандартного раствора хлорида натрия
- § 14. Установка титра 0,1 н. раствора нитрата серебра по точной навеске хлорида натрия
- §15. Определение ионов хлора в техническом хлориде натрия по методу Мора
- § 16. Определение хлоридов по методу Фаянса
- В. РОДАНОМЕТРИЯ
- § 17. Характеристика метода
- § 18. Приготовление 0,1 н. раствора роданида аммония
- § 19. Определение ионов хлора в растворимых хлоридах по методу Фольгарда
- § 20. Определение серебра в сплавах
- Г. МЕРКУРИМЕТРИЯ
- § 21. Характеристика метода
- § 22. Приготовление 0,1 н. раствора нитрата ртути (II)
- § 23. Установка титра раствора нитрата ртути (II)
- § 24. Определение ионов хлора в воде меркуриметрическим методом
- Д. МЕРКУРОМЕТРИЯ
- § 25. Краткая характеристика метода
- Е. КОМПЛЕКСОНОМЕТРИЯ (ХЕЛАТОМЕТРИЯ)
- § 26. Характеристика метода
- § 27. Теоретические основы комплексонометрического титрования
- § 28. Классификация методов комплексонометрического титрования
- § 29. Установка титра раствора комплексона III
- § 30. Определение содержания кальция
- § 31. Определение жесткости воды комплексонометрическим методом
- § 32. Анализ смеси ионов кальция и магния
- § 33. Определение содержания алюминия
- § 34. Раздельное определение ионов кальция и алюминия
- § 35. Раздельное определение ионов алюминия и железа
- Часть вторая. Весовой анализ
- § 1. Сущность весового анализа
- § 2. Классификация методов весового анализа
- § 3. Расчеты в весовом анализе
- Б. ТЕХНИКА ВЕСОВОГО АНАЛИЗА
- § 4. Взятие и растворение навески
- § 5. Техника осаждения
- § 6. Фильтрование и промывание осадков
- § 7. Получение весовой формы
- § 8. Взвешивание весовой формы
- В. ТЕОРЕТИЧЕСКАЯ ЧАСТЬ
- § 9. Теоретические основы выделения осадков из растворов с помощью специфических неорганических и органических реактивов
- § 10. Требования, предъявляемые к осадкам
- § 11. Методы повышения точности весовых определений
- § 12. Теоретические обоснования выбора оптимальных условий для весового определения
- Г. ПРАКТИЧЕСКАЯ ЧАСТЬ
- § 13. Определение кристаллизационной воды в BaCl2 2H2O
- § 14. Определение сульфат-ионов или серы
- § 15. Определение ионов железа (III)
- § 16. Определение содержания кальция в карбонате кальция
- § 17. Определение содержания магния
- § 18. Определение ионов хлора в растворимых хлоридах или в хлористоводородной кислоте
- § 19. Анализ силикатов
- § 20. Анализ доломита
- § 21. Анализ бронзы и латуни
- Д. МЕТОДЫ ВЕСОВЫХ ОПРЕДЕЛЕНИЙ, ОСНОВАННЫЕ НА ПРИМЕНЕНИИ ОРГАНИЧЕСКИХ РЕАКТИВОВ
- § 22. Определение никеля
- § 23. Определение алюминия
- Часть третья. Понятие о физических и физико-химических (инструментальных) методах анализа
- § 1. Электрохимические методы
- § 2. Спектральные (оптические) методы
- § 3. Хроматографические методы
- § 4. Радиометрические методы
- § 5. Масс-спектрометрические методы
- ГЛАВА VII ЭЛЕКТРОВЕСОВЫЕ МЕТОДЫ АНАЛИЗА
- § 1. Характеристика методов электроанализа
- § 2. Химические процессы, протекающие при электролизе
- § 3. Методы электроанализа
- § 4. Электровесовой анализ
- § 5. Метод внутреннего электролиза
- § 6. Определение меди в растворе сульфата меди с применением платиновых сетчатых электродов
- § 7. Определение меди и свинца в латуни с применением платиновых сетчатых электродов
- § 8. Определение малых количеств меди методом внутреннего электролиза
- ГЛАВА VIII. ОБЪЕМНЫЕ ЭЛЕКТРОХИМИЧЕСКИЕ МЕТОДЫ АНАЛИЗА
- § 1. Особенности объемных электрохимических методов анализа
- § 2. Кондуктометрическое титрование
- § 3. Высокочастотное титрование
- § 4. Потенциометрическое титрование
- § 5. Полярографический метод анализа
- § 6. Амперометрическоб титрование
- § 7. Кулонометрическое титрование
- ГЛАВА IX. СПЕКТРАЛЬНЫЕ (ОПТИЧЕСКИЕ) МЕТОДЫ АНАЛИЗА
- § 1. Понятие об эмиссионном спектральном анализе
- Б. КОЛОРИМЕТРИЯ
- § 2. Особенности колориметрических методов анализа
- § 3. Характеристика колориметрических методов анализа
- В. ОПТИЧЕСКИЕ МЕТОДЫ УСТАНОВЛЕНИЯ ТОЧКИ ЭКВИВАЛЕНТНОСТИ
- § 4. Спектрофотометрическое титрование
- § 5. Фототурбидиметрическое и фотонефелометрическое титрование
- Г. ЛАБОРАТОРНЫЕ РАБОТЫ
- § 6. Определение содержания ионов железа методом колориметрического титрования
- § 7. Определение содержания титана
- ГЛАВА X. МЕТОДЫ РАЗДЕЛЕНИЯ, ВЫДЕЛЕНИЯ И КОНЦЕНТРИРОВАНИЯ ОТДЕЛЬНЫХ КОМПОНЕНТОВ АНАЛИЗИРУЕМЫХ СМЕСЕЙ
- § 1. Определение следов элементов (микропримесей)
- § 2. Метод осаждения малорастворимых соединений
- § 3. Электрохимические методы разделения
- § 4. Метод экстрагирования
- § 5. Методы отгонки летучих соединений
- § 6. Хроматографические методы разделения
- § 7. Метод флотации
Ошибки титриметрического анализа также подразделяют на систематические и случайные. Обычно систематические ошибки очень невелики. Причины их будут рассмотрены далее при описании отдельных методов здесь же ограничимся только одним примером. [c.165]
Опшбки в титриметрическом анализе в зависимости от их происхождения могут быть подразделены на методические ошибки, которые связаны с особенностями метода титрования, и специфические ошибки, связанные с особенностями данного метода. [c.349]
Присутствие в растворе белковых веществ и коллоидов, а также нейтральных солей обычно тоже влияет на интервал перехода индикаторов и хотя для титрования применяют лишь те индикаторы, у которых так называемые белковая и солевая ошибки невелики, все же при высоких концентрациях белковых веществ или солей в растворах эти ошибки могут стать значительными. Чтобы исключить влияние всех указанных выше факторов на окончательный результат анализа, каждый раз, когда приходится вести титрование при нагревании или в присутствии неэлектролитов, большого количества солей и т. д., следует устанавливать титр рабочего раствора в тех же самых условиях. Это правило является вообще одним из основных в титриметрическом анализе. [c.253]
Различают прямое титрование, основанное на непосредственном взаимодействии анализируемого вещества и титранта, и обратное титрование, в котором процессу титрования предшествует вспомогательная реакция. Последний метод характеризуется несколько более высокой ошибкой, так как количество измерений при его выполнении возрастает. Для уменьшения суммарной ошибки анализа необходимо, чтобы объем раствора титранта (при выбранной навеске анализируемого вещества) был возможно большим, а ошибка в определении концентрации этого раствора— возможно меньшей. Обычно относительная средняя квадратичная ошибка результатов анализа титриметрическим методом составляет 0,1—0,5%. [c.342]
Ошибки титриметрического анализа подразделяют на систематические и случайные. Обычно систематические ошибки очень невелики. [c.317]
Типичной и наиболее широко распространенной методической погрешностью титриметрических методов анализа является индикаторная ошибка. Она возникает при фиксировании конечной точки титрования. [c.64]
Однонормальные растворы мало пригодны для целей титриметрического анализа как слишком концентрированные. В прибавляемой при титровании ими последней капле раствора содержалось бы, очевидно, довольно много соответствующего вещества, и поэтому так называемая капельная ошибка титрования была бы вел и ка. [c.214]
В титриметрическом анализе часто встречается методическая ошибка, связанная с добавлением избытка реагента по отношению к теоретическому количеству, необходимому для изменения окраски индикатора, по которой судят о конце реакции. В конечном итоге правильность всего анализа определяется спецификой того самого явления, которое лежит в основе определения. [c.60]
Ошибки титриметрического анализа могут быть вызваны следующими причинами. [c.322]
Как будет показано позже, при рассмотрении титрования с внешними индикаторами ошибку, связанную с отбором проб, можно сделать исчезающе малой. Метод равного помутнения, предложенный в 1832 г. Гей-Люссаком, явился одним из первых методов титриметрического анализа. Впоследствии он был нспользован для весьма точного определения атомных весов галогенов и серебра. [c.320]
Каждому методу анализа присущи свои ошибки, которые могут отсутствовать в других методах. Например, ошибки, связанные с потерей вещества при прокаливании, наблюдаются в гравиметрическом анализе, но их нет в титриметрическом анализе. Ошибки, связанные с применением индикаторов, характерны для титриметрического анализа, но отсутствуют в гравиметрическом анализе. Указание на эти ошибки дано при каждом отдельном методе. Есть ошибки, которые характерны для всех методов количественного анализа. Наиример, взвешивая на аналитических весах, можно всегда сделать ошибку, равную 0,0002 г. В тщательно проводимом анализе неорганических веществ относительная ошибка не должна превышать 0,1%. Поэтому навеска вещества для анализа не должна быть меньше 0,2 г. [c.283]
В титриметрическом анализе получаются ошибки при отсчетах на градуированной посуде (бюретках). Например, пользуясь бюреткой на 50 мл, при каждом отсчете мы делаем ошибку 0,02 мл. При отсчете объема в 10 мл ошибка не должна превышать 0,2%. Поэтому надо в случае бюреток на 50 мл работать с объемами растворов не менее 20 мл. [c.283]
Ошибки методические. Эти ошибки зависят от особенностей применяемого метода анализа, например от не вполне количественного протекания реакции, на которой основано определение, от частичной растворимости осадка, от соосаждения вместе с ним различных посторонних примесей, от частичного разложения или улетучивания осадка при прокаливании, от гигроскопичности прокаленного осадка, от течения наряду с основной реакцией каких-либо побочных реакций, искажающих результаты титриметрических определений, от свойств примененного при титровании индикатора и т. д. Методические ошибки составляют наиболее серьезную причину искажения результатов количественных определений, устранить их трудно. [c.48]
Каждому методу анализа свойственны свои специфические ошибки. Например, в гравиметрическом анализе имеют место ошибки, связанные с потерей вещества при промывании и прокаливании осадков. В титриметрическом анализе — ошибки, связанные с применением индикаторов. Наряду с этим имеются ошибки, свойственные всем или многим методам количественного анализа, [c.303]
Операции титриметрического анализа также выполняются с некоторыми, хотя и сравнительно небольшими, ошибками. Считают, что всякое титриметрическое определение включает в себя 1) ошибку определения титра раствора 2) ошибку титрования анализируемого вещества. Первая зависит от точности взвешивания стандартного вещества и правильности измерения объема раствора. Вторгся определяется точностью титрования, т.е. правильностью установления точки эквивалентности с помощью индикатора. Например, если капля раствора, прибавляемая к исследуемой жидкости, имеет слишком большой объем, то этим создается вероятность добавить не строго эквивалентное, а большее количество вещества. [c.165]
Кондуктометрическое титрование расширяет область применения титриметрического анализа, так как благодаря ему становится возможным титрование окрашенных и мутных растворов, когда переход окраски индикатора трудно наблюдать визуально более -точно устанавливается конечная точка при титровании слабых кислот и оснований при кондуктометрическом титровании можно использовать многие реакции осаждения и комплексообразования при анализе смеси веществ повышается точность определений. Относительная ошибка определения находится в пределах 0,1—2% в зависимости от определяемых концентраций. [c.93]
Ошибки в отсчетах по бюретке являются главным источником ошибок в титриметрическом анализе. Особенно часто подобные ошибки допускают начинающие химики, занимая неправильное положение, при отсчете (рис. 92). Относительная ошибка отсчета, вместо допустимого значения 0,1%, может достигнуть 0,3% ли даже 0,6%. [c.109]
Индикаторная ошибка. Своеобразным видом методической ошибки титриметрических методов анализа является индикаторная ошибка. Эта ошибка возникает в связи с тем, что индикатор вступает в реакцию взаимодействия с титрантом либо несколько раньше, либо несколько позже точки эквивалентности. Поскольку взаимодействие определяемого компонента и индикаторного вещества -с титрантом подчиняется законам химического равновесия, момент вступления индикатора в реакцию определяется как прочностью обоих образующихся соединений (константой образования), так и соотношением концентраций искомого компонента и индикатора. Из всех этих величин только концентрация индикатора не является закрепленной и может варьироваться в тех или иных пределах. [c.33]
Другим примером постоянной ошибки служит избыточный объем реагента, необходимый для изменения окраски в титриметрическом анализе. Объем этот обычно мал и не зависит от общего объема реагента, затраченного на титрование. И снова относительная ошибка будет тем выше, чем больше общий объем. Очевидно, одним из путей снижения постоянной ошибки будет выбор разумного количества пробы в соответствии, конечно, с методом анализа. [c.61]
Достоинства потенциометрического метода анализа. Метод потенциометрического титрования имеет при прочих равных условиях ряд достоинств по сравнению с методами визуального титриметрического анализа он более чувствителен, при его использовании исключается субъективная ошибка определения точки эквивалентности. Метод дает возможность проводить определения в мутных и окрашенных растворах и даже в вязких пастах, где невозможно использование индикаторов, и дифференцированно титровать не анализируемые другими методами многокомпонентные смеси веществ без предварительного разделения. [c.43]
Преимущества потенциометрического метода титрования. Потенциометрическое титрование при прочих равных условиях имеет ряд преимуществ по сравнению с визуальными титриметрическими методами анализа. Метод потенциометрического титрования более чувствителен, при использовании его исключается субъективная ошибка, возникающая при визуальном нахождении момента завершения химической реакции, т. е. конечной точки титрования. Этот метод дает возможность определять вещества в мутных и сильно окрашенных растворах, дифференцированно (раздельно) титровать компоненты смесн веществ в одной и той же порции раствора и, наконец, автоматизировать процесс титрования, так как измеряемой величиной является электрический параметр. [c.37]
При определении содержания добавочных компонентов допустима большая ошибка определения [а = 2. .. 5. ..10% (отн.)], особенно при определении небольших содержаний (<10″ %). Вследствие таких требований к точности определения основных и добавочных компонентов для определения первых применяют преимущественно химические методы анализа, для вторых — физико-химические методы. Из химических методов большое применение, благодаря их быстроте, находят титриметрические методы с различными способами определения точки эквивалентности. При особо высоких требованиях к точности прибегают к гравиметрическим методам анализа. Среди физико-химических методов определения добавочных компонентов особенно широкое применение нашли электрохимические методы анализа (полярография, кулонометрия) и оптические (фотометрия). При определении не очень малых количеств элементов (>1%) применяют также различные варианты объемных методов анализа. [c.399]
Систематические ошибки. Систематические ошибки обусловливаются многими причинами. К ним относятся, например, ошибки, зависящие от особенностей метода анализа. Используемые конкретные реакции данного метода могут протекать не вполне количественно, что связано с обратимостью химических процессов. Вместе с осадком могут соосаждаться и посторонние римеси, увеличивая массу осадка. Осадки даже наименее растворимые имеют какую-то частичную растворимость, что уменьшает массу осадка. Осадки могут частично разлагаться при прокаливании, впитывать водяные пары или поглощать газы из атмосферы. В растворе могут происходить побочные реакции. В титриметрических методах некоторые ошибки связаны с используемым индикатором и т. п. К этому типу ошибок относятся ошибки, связанные с личными качествами самого аналитика. Например, не все способны точно уловить момент перемены окраски раствора в процессе титрования, не всегда точно улавливаются мелкие деления на шкале приборов. Иногда [c.215]
В вычислении результатов титриметрических определений наименее точная цифра — число миллилитров титрующего раствора, израсходованного на титрование. Поскольку сотые доли миллилитра отмечаются лишь приблизительно, можно принять, что максимальная ошибка отмеривания не менее 0,02 мл. Ошибка от натекания также равна 0,02 мл. Таким образом, общая ошибка может доходить до 0,04 мл . При общем расходе титрующего раствора 20 мл это составит 0,2% отн. Отсюда следует, что, беря для анализа 1 г, вполне можно проводить отвешивание с точностью до 1 мг это дает относительную ошибку в 0,5 мг, или 0,05%. Если на титрование расходуется меньше 20 мл [c.11]
Ошибки эталонов и стандартов. Определение содержания отдельных компонентов во многих методах химического анализа опосредовано через применение разного рода стандартов и эталонов. Таковы методы фотометрического, эмиссионного спектрального, атомно-абсорбционного, газохроматографического анализов, полярографические, амперометрические, кон-дуктометрические, радиохимические и многие другие методы. В последнее время в титриметрических методах получили [c.35]
Преимущества метода кулонометрического титрования также ощутимы в тех случаях, когда для проведения анализа требуется малое количество реагента. Регулируя силу тока, можно легко и с высокой точностью вводить в раствор небольшие порции реагента, тогда как в классических титриметрических методах дозирование малых объемов даже сильно разбавленных растворов приводит к значительным ошибкам. [c.43]
Систематические ошибки. Систематические ошибки обусловливаются многими причинами. К ним относятся, например, ошибки, зависящие от особенностей метода анализа. Используемые конкретные реакции данного метода могут протекать не вполне количественно, что связано с обратимостью химических процессов. Вместе с осадком могут соосаждаться и посторонние примеси, увеличивая массу осадка. Осадки даже наименее растворимые имеют какую-то частичную растворимость, что уменьшает массу осадка. Осадки могут частично разлагаться при прокаливании, могут впитывать водяные пары или поглощать газы из атмосферы. В растворе могут происходить побочные реакции. В титриметрических методах ошибки могут быть связаны с используемым индикатором и т. п. К этому типу ошибок относятся ошибки, связанные с личными качествами самого аналитика. Например, не все могут точно уловить момент перемены окраски раствора в процессе титрования, не всегда точно улавливаются мелкие деления на шкале приборов. Иногда учащиеся ориентируются на определенные данные или на цифры, которые получают работающие рядом, а не на свои собственные. Это так называемые психологические ошибки, зависящие от настроения самого исполнителя. [c.227]
Малые количества углекислоты (несколько ч. на млн.) определить очень трудно, так как в воздухе содержится значительное количество СО2. Поэтому химический анализ связан с трудностью определения поправки холостого опыта. Вследствие того, что аммиак и С02 образуют химическое соединение, обычные методы газового анализа «здесь оказываются неприемлемыми. В работе [443] предложен титриметрический метод определения СО2 в безводном аммиаке, основанный па связывании углекислоты в виде карбамата аммония, отделении аммиака отгонкой при комнатной температуре, обработке остатка разбавленной серной кислотой, поглощении выделяющейся СО2 гидроокисью бария и титровании избытка последнего щавелевой кислотой. Этим методом при содержании углекислоты 0—50 ч. на млн. абсолютная ошибка равна 1,5 ч. на млн. [c.257]
На результат анализа могут оказывать влияние и случайные ошибки. Чтобы устранить их, титриметрическое определение повторяют несколько раз и берут среднее из них. Однако, вычисляя средний результат, допускают отклонения не более 0,3%. Результаты определений, отличающиеся на большую величину, отбрасывают при вычислении среднего арифметического. Например, если в четырех определениях нормальности раствора едкого натра были получены величины 0,1134, 0,1135, 0,1142 и 0,1136, то число 0,1142 отбрасывают, оно отличается от наименьшего числа 0,1134 на 0,8%. Из остальных величин берут среднее арифметическое. [c.318]
Весьма важное значение имеет правильное вычисление результатов титриметрического анализа. Все вычисления рекомендуется выполнять со всей тщательностью и внимательно, так как правильные результаты титрования из-за ошибок в расчетах дают неверный результат. Всякое определение включает два рода ошибок ошибку в концентрации титрующего раствора и ошибки титрования определяемого вещества. Эти ошибки компенсируются в том случае, если концентрация титруюшего раствора установлена в тех же условиях, что и титрование анализируемого образца. Влияние случайных ошибок можно устранить, повторяя титрование несколько раз. Отклонение от среднего результата не должно превышать 0,3% относительных. Поэтому отсчеты объемов по бюретке необходимо вести с точностью до 0,02— 0,03 мл. Например, три последовательных титрования дают 25,06 25,03 25,03 мл. Средний результат титрования должен быть записан в виде 25,04 мл. Если отклонения превышают допустимую величину, то такие результаты титрования не должны приниматься во внимание при вычислении среднего результата. Для повышения точности измерения объема применяют бюретки малого диаметра или весовые бюретки. [c.351]
Проведенный выше раэбор систематических ошибок хими-t e Koro аяализа не претендует на исчерпывающую полноту. Из рассмотрения исключены некоторые виды ошибок, например, ошибка натекания и капельная ошибка в титриметрических методах анализа. Некоторые виды систематических ошибок только упомянуты. Основное внимание и наибольшее количество примеров посвящено ошибкам традиционных методов гравиметрического, титриметрического и фотометрического анализов. Такой стиль изложения оправдан целью данного раздела—дать общее представление о систематических ошибках химического анализа, способах их обнаружения и оценки и методах их уменьшения. Детальный разбор всех известных источников ошибок должен входить как составная часть в теорию и практику каждого отдельного метода химического анализа, ибо каждому методу присущи свои специфические ошибки». Удачным примером в этом плане может служить руководство по (фотоколориметрическим и спектрофотометрическим методам анализа М. И. Булатова и И. П. Калин-кина (Л, Химия , 1976, 376 с.), где этому вопросу уделено большое внимание. Однако сказанное в равной мере относится и к любым другим химическим и физическим методам, [c.48]
В качестве титруюш его раствора при аналогичном определении может быть использован метапериодат [1176]. В работе [59] проведено титриметрическое определение ртути нри осаждении ее перйодатом калия в виде Hg5(100)2, растворении осадка в кислоте и иодометрическим титровании. При определении от 0,2 до 0,02 г Hg(II) ошибка равна +0,2%. Для анализа соединений Hg(II) можно также использовать данный метод, предварительно восстановив Hg(II) до Hg(I). [c.90]
Элементный бром в водах определяют титрованием раствором соли Мора по N,N-диэтил- г-фeнилeндиaминy [739, 741] согласно описанию на с. 76. Для его определения в водных растворах рекомендованы экстракционные методы с фотометрическим и титриметрическим окончанием [157]. Они требуют небольшого расхода времени, но ошибки анализа достигают 20—50%. Для качественного определения брома можно применять реакции, рассмотренные в главе III. [c.179]
Для анализа производственных вод применяется титриметрический метод определения сульфидов и цианидов потенциометрическим титрованием, разработанный в ВУХИНе [ 1 ]. Данный метод значительно сокращает время анализа, количество реактивов. Время определения составляет 10—15 мин, ошибка — 1 — 2 % (отн.) (химические методы требуют [c.59]
К ошибкам этого же типа в значительной мере можно отнести также индикаторные ошибки в титриметрических методах, поскольку во многих случаях их можно предрассчитать. В какой-то мере возможна приближенная оценка отдельных видов методической ошибки и в других методах химического анализа. Однако обычно методические ошибки включают в себя отдельные слагаемые, обусловленные не только неполнотой протекания равновесных процессов или параллельным протеканием сопроцессов , но и неравновесностью отдельных стадий химико-аналитических процессов, т. е. связанные с кинетическими факторами. [c.25]
При титровании следовых количеств хлорид-ионов возможна ошибка за счет реакции НС1 с СО,. Такую ошибку можно корректировать введением поправки [720]. Ошибку титрования увеличивает скоагулировавшийся осадок. Поэтому для предотвращения коагуляции к концентрированным растворам хлоридов лития,, натрия, калия, магния, кальция добавляют 5—10 мл 0,1%-ного раствора агар-агара. В качестве титранта используют 0,01— 0,1 N AgNOg (чаще всего 0,05А ). Метод Мора применяют в основном для анализа вод [633, 771]. В настоящее время он все больше вытесняется другими титриметрическими методами, исключающими использование серебра. Ограничивает также применимость этого метода необходимость проведения анализа в нейтральной срвде, что исключает присутствие в ана.иизируемых объектах тяжелых металлов. Поэтому гораздо чаще применяют метод Фоль-гарда, позволяющий проводить титрование в кислой среде. [c.36]
Большинство методов количественного ультрамикрохимического анализа основано на измерении какого-либо параметра, функционально связанного с массой. Это, прежде, всего, титриметрические и фотометрические методы. Важным преимуществом физико-химических методов является то, что при их использовании нет необходимости брать навески на высоко точных и чувствительных ультрамикровесах, так как микронавеска может быть взята и отбором аликвотной части раствора. При анализе жидкостей измеряют объем, взятый для анализа. В физических методах, как, например, эмиссионном спектральном, рентгеноспектральном, масс-спектральном анализе навески нЗ микровесах вообще не берут. Конечно, и в этих методах точность анализа зависит во многом от погрешности приборов. Во всяком случае в ультрамикроанализе, так же как и в микроанализе, случайные ошибки приборов составляют, по крайней мере, треть погрешности, вызываемой химическими факторами [c.183]
Анализ сплавов никеля. Предложено несколько методов, в зависимости от определяемых количеств вольфрама. Титриметрический метод [746] применяют для определения 0,5—5% W из навески 0,1 г сначала восстанавливают W(VI) до W(III) свинцом, затем вводят избыток Fe(III), восстановленное железо оттитровывают К2СГ2О7. Для определения 0,04—5% W разработан [153] экстракционно-фотометрический метод с помощью роданида и хлорида N,N N»-тpифeнилгyaнидиния. При определении 0,05 — 1,0% W и 1—5% W ошибка определения составляет соответственно + 10% и + 2—5%. Флуориметрический метод [561] позволяет определять 0,5—3% W из навески 0,05—0,1 г. [c.178]
Для определения сантимиллиграммовых количеств азота в органических соединениях предложен титриметрический метод, состоящий в разложении вещества серной кислотой в запаянной кварцевой трубке при нагревании, пропускании реакционной смеси через ионообменную смолу основного характера и титровании образовавшегося NH4OH раствором HGIO4 с потенциометрической регистрацией КТТ [782]. Продолжительность анализа 12 мин. Определяемый минимум 30 мкг N. Абсолютная ошибка +0,25%. Металлы мешают определению. [c.182]
Курс аналитической химии Издание 5 (1982) — [
c.98
,
c.295
]
Курс аналитической химии Кн 2 Издание 4 (1975) — [
c.98
,
c.301
]
В университете на практическом курсе аналитической химии мне приходилось много титровать. Я помню осторожное дозирование титранта стеклянной бюреткой, неудобный процесс повторного наполнения бюретки и постоянное опасение, что я неправильно выбрала конечную точку.
Все студенты в группе получали разные результаты, но мы так и не поняли почему. В те годы у меня было мало знаний. Сейчас, после 10 лет опыта титрования, я поняла, что результаты ручного титрования очень сильно зависят от человека, который проводит анализ. Ниже я перечислю основные источники ошибок, и как вы можете их избежать.

Правильный выбор индикатора
Значение pH конечной точки титрования зависит от константы кислотной диссоциации (Ka) используемых кислоты и основания. Если сильное основание титруют сильной кислотой, значение pH в конечной точке составляет около 7. Титрование сильного основания слабой кислотой смещает конечную точку в сторону диапазона щелочи. Титрование сильной кислоты слабым основанием – в зону кислот. Это объясняет, почему при кислотно-щелочном титровании используются несколько разных индикаторов. Какой мы выберем?
На приведенной выше диаграмме показаны самые популярные pH индикаторы. Из графика видно, что не получите правильных результатов при pH конечной точки = 7, если в качестве индикатора вы используете кристаллический фиолетовый или метиловый оранжевый. К счастью, в большинстве стандартов и в стандартных методиках работы индикатор уже указан. Если будете следовать инструкциям, все будет хорошо!

Субъективность распознавания конечной точки
Проблемы начинаются в процессе определения конечной точки. Вы когда-нибудь задумывались о тонкостях изменения цвета?
На рисунке вы видите пять стадий кислотно-щелочного титрования c(HCl) = 1 моль/л с c(NaOH) = 1 моль/л. Единственная разница между соседними фотографиями заключается в одной капле титранта. На каком из них истинная конечная точка?
Может это конечная точка на рисунке 1 с бледно-розовым оттенком? Или на картинке под номером 3 с более интенсивным цветом? Или даже на картинке 5, где розовый цвет наиболее яркий? Между 1 и 5 было добавлено всего четыре капли титранта. Одна капля – это 50 мкл, а добавленный в сумме объем составляет 200 мкл титранта или примерно 7,3 мг соляной кислоты. Для фармацевтического анализа – это колоссальная ошибка.

Считываем объем на бюретке
Вы помните, как правильно считывать значения на бюретке? Нужно убедиться, что фиксируете значение мениска четко по горизонтали. Знаете почему?
Значение объема зависит от угла, под которым вы смотрите на бюретку. На данном рисунке данные отличаются от фактического значения до 0,2 мл (200 мкл) в зависимости от угла обзора. Чем больше ваш ракурс отклоняется от горизонтали, тем более неточными будут значения и сам результат. Вы можете допустить среднюю ошибку 200 мкл. Но для титрования это недопустимо много, на предыдущем примере я рассказала почему.
Повышаем объективность и точность оценки
Как можно исключить эти ошибки? Легче всего просто обойти возможную неточность. Решением станет использование электронной бюретки. Все, что вам нужно сделать, это заполнить ее титрантом, а затем нажать кнопку. Устройство автоматически измерит объем и выдаст оцифрованный результат. Использование электронной бюретки уже обеспечит высокий уровень объективности ваших результатов.
Электронная бюретка также повысит и точность данных. Мне не нужно рассказывать вам, насколько важна точность в аналитической химии, но я приведу пример. Представьте, что вы определили чистоту золота в 90%, а на самом деле оно чистое на 99%. Вы бы потеряли много денег, продавая свое золото с этой иллюзией!
Ранее я показала, что визуальное распознавание конечной точки с цветовым индикаторам может привести к ошибкам до 200 мкл. Неточность ручной бюретки может вызвать погрешность в дополнительных 200 мкл. Хотя электронная бюретка не помогает добиться лучшей объективизации при распознавании конечной точки, она добавляет куда меньший объем титранта: вместо прежних 50 мкл на каплю, всего 0,25 мкл в зависимости от объема используемой бюретки. Это существенно снизит ошибку при распознавании конечной точки. Самые распространенные бюретки описаны ниже:
| Объем цилиндра бюретки (мл) | Мин. добавляемый объем (µл) |
| 5 | 0.25 |
| 10 | 0.50 |
| 20 | 1.00 |
| 50 | 2.50 |
Следующий шаг: автоматическое титрование
Если вы хотите закрыть все источники ошибок, про которые мы говорили, вам придется перейти на автоматическое титрование или автотитрование. В этом случае вы будете использовать электрод для анализа изменения pH в образце и математический алгоритм для детектирования конечной точки — индикатор вам больше не потребуется. Кроме того, вы получите ту же точность, что и с электронной бюреткой.
Ознакомьтесь с соответствующей записью в блоге для получения дополнительной информации.
How to Transfer Manual Titration to Autotitration
Author

Iris Kalkman
Product Specialist TitrationMetrohm International Headquarters, Herisau, Switzerland