Текст ГОСТ Р 58578-2019 Правила установления нормативов и контроля выбросов запаха в атмосферу
ГОСТ Р 58578-2019
НАЦИОНАЛЬНЫЙ СТАНДАРТ РОССИЙСКОЙ ФЕДЕРАЦИИ
ПРАВИЛА УСТАНОВЛЕНИЯ НОРМАТИВОВ И КОНТРОЛЯ ВЫБРОСОВ ЗАПАХА В АТМОСФЕРУ
Regulations for establishing environmental standards for odour and performing control of odour emissions
ОКС 01.040.13
Дата введения 2020-01-01
Предисловие
1 РАЗРАБОТАН Акционерным обществом «Научно-исследовательский институт охраны атмосферного воздуха» (АО «НИИ Атмосфера»)
2 ВНЕСЕН Техническим комитетом по стандартизации ТК 457 «Качество воздуха»
3 УТВЕРЖДЕН И ВВЕДЕН В ДЕЙСТВИЕ Приказом Федерального агентства по техническому регулированию и метрологии от 8 октября 2019 г. N 889-ст
4 Настоящий стандарт соответствует европейскому стандарту ЕН 13725:2003 «Качество воздуха. Определение концентраций запахов методом динамической ольфактометрии» (EN 13725:2003 «Air quality — Determination of the odour concentration by dynamic olfactometry», NEQ) в части требований установления нормативов и контроля выбросов запаха в атмосферу
________________
* Доступ к международным и зарубежным документам, упомянутым в тексте, можно получить, обратившись в Службу поддержки пользователей. — .
5 ВВЕДЕН ВПЕРВЫЕ
Правила применения настоящего стандарта установлены в статье 26 Федерального закона от 29 июня 2015 г. N 162-ФЗ «О стандартизации в Российской Федерации«. Информация об изменениях к настоящему стандарту публикуется в ежегодном (по состоянию на 1 января текущего года) информационном указателе «Национальные стандарты», а официальный текст изменений и поправок — в ежемесячном информационном указателе «Национальные стандарты». В случае пересмотра (замены) или отмены настоящего стандарта соответствующее уведомление будет опубликовано в ближайшем выпуске ежемесячного информационного указателя «Национальные стандарты». Соответствующая информация, уведомление и тексты размещаются также в информационной системе общего пользования — на официальном сайте Федерального агентства по техническому регулированию и метрологии в сети Интернет (www.gost.ru)
Введение
Рефлекторное воздействие на человека индивидуального вещества, обладающего запахом, учитывается при установлении предельно допустимой концентрации (ПДК) данного вещества. В большинстве случаев запах формируется не отдельным веществом, а сложной смесью веществ, из состава которой часто невозможно выделить конкретные обладающие запахом соединения, большинство из которых не идентифицированы и не имеют ПДК. Кроме того, даже те пахучие соединения в смеси, для которых установлен норматив ПДК, часто присутствуют в атмосферном воздухе в таких незначительных количествах, что при контроле качества атмосферного воздуха превышение ПДК несмотря на наличие отчетливого запаха, как правило, не наблюдается.
Когда запах формируется не индивидуальным веществом, а смесью пахучих веществ неизвестного состава, осуществляют контроль не за выбросами отдельных пахучих веществ, а контролируют запах в целом. Мероприятия по контролю запаха включают самую разнообразную деятельность, в том числе опросы населения, анализ поступающих от населения жалоб, инспекторские проверки и т.д. Однако полную количественную оценку запаха в воздухе или выбросах предприятия могут дать только ольфактометрические исследования запаха, а также последующее моделирование распространения выбросов запаха в атмосфере.
При установлении гигиенических нормативов для индивидуальных веществ, обладающих сильным запахом, учитывают не только их непосредственное влияние на здоровье, но и раздражающее воздействие запаха на психическое состояние человека. Однако ощущение запаха чаще всего создается не одним конкретным веществом, а смесью пахучих веществ переменчивого состава. Выделение из такого рода смеси индивидуальных веществ и их нормирование в большинстве случаев является необычайно трудоемким и нецелесообразным.
Гигиенические нормативы индивидуальных пахучих веществ, а также некоторых смесей пахучих веществ (например, ПДК для летучих компонентов ароматизаторов, применяемых в производстве жевательной резинки, и ПДК для летучих компонентов смеси душистых веществ и эфирных масел, содержащихся в выбросах предприятий парфюмерно-косметической промышленности) установлены с учетом их рефлекторного воздействия на человека и выражены в миллиграммах на кубический метр.
В настоящее время в России в целом отсутствует система нормирования запаха в атмосферном воздухе. При наличии источников, выбрасывающих пахучие вещества, постоянные жалобы населения на неприятный запах вынуждают местные органы власти предпринимать всевозможные меры и, в частности, проводить исследования выбросов предприятий. В то же время подобные исследования при отсутствии нормативной базы в отношении запаха имеют ограниченную область применения и не позволяют природоохранным органам воздействовать на предприятия и требовать проведения мероприятий по снижению выбросов пахучих веществ.
В настоящем стандарте предлагаются некоторые подходы к развитию нормирования запаха, учитывающие как отечественные методы гигиенического нормирования, так и зарубежную практику.
При разработке системы нормирования запаха применяют тот же подход, который используют для нормирования пахучих веществ. При этом необходимо отметить, что ПДК пахучего вещества выражена в миллиграммах на кубический метр, тогда как норматив запаха будет иметь размерность ЕЗ/м, где ЕЗ (единица запаха) в кубическом метре воздуха представляет собой концентрацию запаха, которую ощущает половина экспертов, принимающих участие в исследовании.
Апробирование на конкретных примерах вышеописанного способа установления ПДК пахучих веществ для определения норматива запаха в атмосферном воздухе (ЕЗ/м) показало, что использование величины
![]() , соответствующей 16% обнаружения запаха, приводит к очень низким нормативным значениям концентрации запаха в атмосферном воздухе (менее 1 ЕЗ/м
, соответствующей 16% обнаружения запаха, приводит к очень низким нормативным значениям концентрации запаха в атмосферном воздухе (менее 1 ЕЗ/м). Такие низкие нормативы запаха не применяются в других странах, так как являются труднодостижимыми и практически не поддаются контролю. Поэтому для определения норматива запаха целесообразно использовать не 16%, а большее значение, выбор которого будет также зависеть от интенсивности и гедонического тона запаха, а также места расположения и параметров источника выброса запаха.
Аналогично следует устанавливать местные, локальные нормативы запаха, действующие на относительно небольшой территории вблизи предприятия, выбросы пахучих веществ которого негативно воздействуют на население. При установлении нормативного значения помимо учета таких факторов, как плотность населения и значимость предприятия для данного региона, также целесообразно проводить анализ жалоб населения на неприятный запах с учетом метеорологических факторов и результатов рассеивания выбросов. Сопоставляя результаты расчета и места проживания населения, из которых поступает основной поток жалоб, можно определить зону воздействия выбросов запаха на людей и скорректировать рассчитанный норматив запаха таким образом, чтобы уменьшить количество жалоб от населения, проживающего за пределами изолинии нормативной концентрации.
1 Область применения
Настоящий стандарт устанавливает правила контроля выбросов запаха в атмосферу.
Настоящий стандарт распространяется на методы исследования запаха в выбросах промышленных предприятий и в атмосферном воздухе, требования к измерению концентрации запаха ольфактометрическим способом, оборудование и материалы, используемые в ольфактометрии.
2 Нормативные ссылки
В настоящем стандарте использованы нормативные ссылки на следующие стандарты:
ГОСТ 17.2.3.02-2014 Правила установления допустимых выбросов загрязняющих веществ промышленными предприятиями
ГОСТ 17.2.4.06 Охрана природы. Атмосфера. Методы определения скорости и расхода газопылевых потоков, отходящих от стационарных источников загрязнения
ГОСТ 17.2.4.07 Охрана природы. Атмосфера. Методы определения давления и температуры газопылевых потоков, отходящих от стационарных источников загрязнения
ГОСТ 17.2.4.08 Охрана природы. Атмосфера. Методы определения влажности газопылевых потоков, отходящих от стационарных источников загрязнения
ГОСТ ISO 5492 Органолептический анализ. Словарь
ГОСТ ISO 8589 Органолептический анализ. Общее руководство по проектированию лабораторных помещений
ГОСТ ISO 16000-30 Воздух замкнутых помещений. Часть 30. Органолептический анализ воздуха замкнутых помещений
ГОСТ Р 8.736 Государственная система обеспечения единства измерений. Измерения прямые многократные. Методы обработки результатов измерений. Основные положения
ГОСТ Р 53701 Руководство по применению ГОСТ Р ИСО/МЭК 17025 в лабораториях, применяющих органолептический анализ
Примечание — При пользовании настоящим стандартом целесообразно проверить действие ссылочных стандартов в информационной системе общего пользования — на официальном сайте Федерального агентства по техническому регулированию и метрологии в сети Интернет или по ежегодному информационному указателю «Национальные стандарты», который опубликован по состоянию на 1 января текущего года, и по выпускам ежемесячного информационного указателя «Национальные стандарты» за текущий год. Если заменен ссылочный стандарт, на который дана недатированная ссылка, то рекомендуется использовать действующую версию этого стандарта с учетом всех внесенных в данную версию изменений. Если заменен ссылочный стандарт, на который дана датированная ссылка, то рекомендуется использовать версию этого стандарта с указанным выше годом утверждения (принятия). Если после утверждения настоящего стандарта в ссылочный стандарт, на который дана датированная ссылка, внесено изменение, затрагивающее положение, на которое дана ссылка, то это положение рекомендуется применять без учета данного изменения. Если ссылочный стандарт отменен без замены, то положение, в котором дана ссылка на него, рекомендуется применять в части, не затрагивающей эту ссылку.
3 Термины и определения
В настоящем стандарте применены термины и определения согласно приведенным в нормативном документе, а также следующие термины с соответствующими определениями:
_______________
См. [1].
3.1 европейская единица запаха: Количество пахучего вещества (пахучих веществ), которое(ые), будучи разбавленным(и) 1 м нейтрального газа при стандартных условиях (температура 20°С и давление 101,3 кПа), вызывает физиологический отклик, эквивалентный отклику, вызываемому одной европейской эталонной массой запаха (EROM), разбавленной 1 м
нейтрального газа при стандартных условиях.
3.2 европейская эталонная масса запаха; EROM (European Reference Odour Mass): Определенная масса аттестованного стандартного образца.
Примечание — Один EROM эквивалентен 123 g н-бутанола (CAS-Nr. 71-36-3). Испаренный в одном кубическом метре нейтрального газа — эквивалентен концентрации 0,040
mol/mol.
3.3 единица запаха, ЕЗ/м: Концентрация запаха, которую ощущают 50% испытателей; количество (смеси) одорантов в 1 м
пахучего газа (при температуре 20°С и давлении 101,3 кПа) при достижении порога выявления экспертной комиссии.
3.4 интенсивность запаха: Количественная характеристика запаха, которая показывает, насколько сильным кажется человеку воспринимаемый запах.
3.5 коэффициент разбавления: Соотношение между объемом пробы после ее разбавления нейтральным газом и объемом пахучего газа.
3.6 метод «да/нет»: Ольфактометрический метод, при котором экспертов просят зафиксировать наличие запаха или его отсутствие.
3.7 метод принудительного выбора: Ольфактометрический метод, при котором эксперты вынуждены выбирать из двух потоков воздуха или более, один из которых представляет собой разбавленную пробу, даже в том случае, если не наблюдается ощутимой разницы между потоками.
3.8 нейтральный газ: Воздух или азот, который подготавливается таким образом, чтобы из него по возможности были удалены все запахи, и который, по мнению членов экспертной группы, не мешает проведению исследования конкретного запаха.
3.9 ольфактометрия: Метод измерения запаха по степени его воздействия на испытуемого при помощи специальных приборов ольфактометров.
3.10 ольфактометр: Прибор, в котором проба пахучего газа подвергается разбавлению нейтральным газом (в определенном соотношении) и представляется экспертам для анализа.
3.11 оценочное значение индивидуального порога; ITE: Порог выявления применительно к одному эксперту, рассчитанный на основе серии разбавлений.
3.12 пахучее вещество: Вещество, которое воздействует на обонятельную систему человека в такой степени, что человек чувствует запах.
3.13 порог выявления: Значение разбавления, при котором вероятность выявления запаха в условиях исследования составляет 50%.
3.14 порог экспертной комиссии: Порог выявления применительно к экспертной группе.
3.15 предельно допустимая концентрация; ПДК: Максимальная концентрация химических элементов и их соединений в окружающей среде, которая при повседневном влиянии в течение длительного времени на организм человека не вызывает патологических изменений или заболеваний, устанавливаемых современными методами исследований, в любые сроки жизни настоящего и последующего поколений.
3.16 представление: Представление одному эксперту для оценки запаха одной смеси в виде разбавленной пробы запаха или нейтрального газа.
3.17 проба: Проба газовоздушной смеси или атмосферного воздуха, содержащая пахучие вещества в количестве, формирующем запах.
3.18 разбавление: Процесс смешения двух известных потоков газа, а именно — пробы пахучего вещества и нейтрального газа.
Примечание — Степень разбавления рассчитывают, исходя из показателей расхода потока.
3.19 разбавление до порогового уровня: Показатель числа разбавлений, которое необходимо для того, чтобы изначально пахучий атмосферный воздух стал «неопределимым», т.е. нос человека (эксперта) перестал ощущать запах.
3.20 раунд: Представление одной серии проб всем экспертам.
3.21 серия представлений: Представление всем членам экспертной группы одной смеси за один раунд.
3.22 серия разбавлений: Предъявление последовательности разбавлений [в случайном порядке или в порядке увеличения концентрации запаха (уменьшения разбавления)] одному члену экспертной группы.
3.23 стандартные условия для ольфактометрии: Комнатная температура 20°С и атмосферное давление 101,3 кПа.
3.24 экспертная группа по запахам: Группа экспертов, участвующих в процессе исследования запаха, все члены которой удовлетворяют определенным критериям отбора.
3.25 эксперт по запахам: Лицо, участвующее в исследовании запаха, удовлетворяющее определенным условиям отбора экспертов.
4 Основания для проведения исследований запаха на предприятии
Основаниями для проведения исследований запаха на предприятии являются:
— жалобы населения на неприятный запах;
— предписания органов исполнительной власти, осуществляющих контроль атмосферного воздуха;
— желание руководства предприятия.
5 Методы исследования запаха
5.1 Метод исследования запаха на источнике выбросов
Метод исследования на источнике включает отбор и ольфактометрический анализ проб запаха для определения концентрации запаха в выбросах и последующий расчет рассеивания выбросов запаха в атмосферном воздухе с целью оценки уровня воздействия запаха на население. Метод позволяет получить количественные данные, на основе которых разрабатывают нормативы допустимого уровня воздействия.
Количественное определение запаха осуществляют с применением ольфактометрии, представляющей собой количественный метод измерения запаха по степени его воздействия на человека. Цель измерения запаха заключается в определении степени дискомфорта, вызванного запахом. При исследовании методом ольфактометрии присутствие и интенсивность запаха оценивает группа экспертов с учетом воздействия на их органы обоняния. Обоняние человека характеризуется высокой чувствительностью и по-разному реагирует на разнообразные химические вещества. Порог восприятия запаха человеком — это такая концентрация пахучих веществ в воздухе, при превышении которой человек способен почувствовать запах и которая в значительной степени зависит от природы вещества и может колебаться в широких пределах. Обоняние реагирует даже на концентрацию определенных пахучих веществ, которая недоступна для обнаружения инструментальными методами. Поэтому искусственные детекторы не способны по результатам измерений определить воздействие конкретных веществ на обоняние человека, так как во многих случаях их чувствительности недостаточно.
В ольфактометрии в качестве детектора измерения запаха применяют орган обоняния человека — нос. Прибор для измерения запаха с помощью человеческого носа называется ольфактометром. Ольфактометр — это устройство, в котором пахучее вещество разбавляется чистым воздухом в разных пропорциях и, соответственно, в разных концентрациях подается членам экспертной группы для оценки. Измерения могут проходить в порядке увеличения концентрации, начиная с концентраций ниже порога восприятия (предельный метод) или в случайной последовательности выше и ниже порога восприятия (непрерывный метод). Эксперт должен выбрать один из ответов: «да, пахнет» или «нет, не пахнет». С помощью ольфактометра, понижая степень разбавления вещества воздухом, измеряют порог восприятия (одну единицу запаха в 1 м), т.е. такую концентрацию запаха, которую способны воспринимать как запах 50% испытуемых. Измерения запаха на ольфактометре позволяют получить его концентрацию в единицах запаха (ЕЗ/м
).
При применении метода принудительного выбора воздух для оценки подается эксперту поочередно через два порта ольфактометра или более, при этом по одному из портов поступает пахучая проба в том или ином разбавлении, а по другому порту (или по остальным портам) — нейтральный газ. Выбор порта для подачи пахучей пробы осуществляют в случайном порядке для каждого представления. В каждом представлении эксперт должен выбрать порт с пахучей пробой; если разница не ощущается, эксперт вынужден выбирать «наугад». Для каждого ответа эксперт указывает степень своей уверенности в соответствии с таблицей 1, после чего все ответы обрабатывают.
Таблица 1 — Реакция экспертов при применении метода принудительного выбора
|
Ответ |
Результат выбора |
Степень уверенности |
|
Ложь |
Неверный |
Догадка |
|
Ложь |
Верный |
Догадка |
|
Ложь |
Неверный |
Легкое подозрение |
|
Ложь |
Верный |
Легкое подозрение |
|
Ложь |
Неверный |
Уверенность |
|
Истина |
Верный |
Уверенность |
Оценочное значение индивидуального порога рассчитывают как среднее геометрическое между первым из двух последовательных разбавлений, для которых получены истинные ответы, и предыдущим (большим) разбавлением, для которого получен ложный ответ. Данный расчет может быть выполнен и программой ольфактометра автоматически.
Полученное значение концентрации используют для расчета мощности выброса запаха и дальнейшего моделирования распространения запаха в атмосфере. Запах, исходящий от исследуемых источников выбросов, связан с поступлением в атмосферу разнообразных пахучих веществ, совокупные выбросы которых и создают неприятные для человека ощущения. Запах формируется загрязняющим веществом или смесью загрязняющих веществ, закономерности переноса вещества и запаха в атмосферном воздухе являются одинаковыми.
Для изучения распространения запаха в атмосфере можно применять те же математические модели, что и для расчета рассеивания выбросов загрязняющих веществ. В настоящее время в Российской Федерации используются «Методы расчетов рассеивания выбросов вредных (загрязняющих) веществ в атмосферном воздухе» [2].
Метод исследований запаха на источнике выбросов пахучих веществ позволяет:
— получать объективные количественные данные о выбросах запаха от конкретной установки или технологического процесса;
— оценивать эффективность мероприятий по снижению выбросов пахучих веществ;
— рассчитывать формируемые в атмосферном воздухе концентрации запаха в окрестностях предприятия — источника неприятного запаха.
5.2 Метод полевых исследований запаха в окрестностях предприятия
5.2.1 Измерение запаха на местности
Метод полевых исследований позволяет оценить запах непосредственно в выбранных точках на местности вблизи источника выбросов.
Для оценки частоты возникновения запаха как показателя уровня воздействия зона исследования разбивается на квадраты. Во время проведения исследований члены экспертной группы должны посетить каждый из углов квадрата более или менее равномерно в течение всего периода проведения измерений (не менее 6 мес). Длительность каждого из наблюдений составляет 10 мин, в течение которых регистрируют присутствие и характер запаха. Мониторинг запаха в каждой точке следует проводить несколько раз в неделю (частота измерений может варьироваться в зависимости от целей мониторинга). Фиксируют все присутствующие запахи, при этом обязательно указывают их характер. При дальнейшем анализе всех собранных данных все случаи наличия запаха ретроспективно разделяют на непосредственно связанные с работой исследуемого предприятия (источника выбросов запаха) и запахи, которые ни при каких обстоятельствах не могут быть обусловлены основной деятельностью предприятия. Последние исключаются из дальнейшего рассмотрения.
Более простой способ оценки запаха на местности — это метод подфакельных наблюдений, используемый для выявления зон распространения загрязняющих веществ от источника выбросов. Места оценки запаха при подфакельных наблюдениях выбирают на разных расстояниях от источника загрязнения с учетом закономерностей распространения загрязняющих веществ в атмосфере. Точки исследования запаха располагают последовательно по направлению ветра на определенных расстояниях от источника выброса, а также с наветренной стороны источника согласно ГОСТ ISO 5492, ГОСТ ISO 16000-30.
5.2.2 Использование переносных ольфактометров
Полевые исследования запаха в контрольных точках осуществляют с помощью переносных ольфактометров, которые позволяют проводить количественные измерения концентрации запаха в атмосферном воздухе. Принцип действия переносных (ручных) ольфактометров, выпускаемых в настоящее время, заключается в смешивании пахучего атмосферного воздуха с отфильтрованным воздухом, не содержащим примесей, или разбавлении пахучего атмосферного воздуха чистым воздухом из баллона.
В первом случае группа экспертов оценивает наличие запаха в пробах, содержащих загрязненный воздух, который постепенно разбавляют очищенным воздухом до уровня, при котором эксперты не ощущают его. С помощью прибора проводят несколько серий дискретных разбавлений загрязненного запахом атмосферного воздуха чистым воздухом. Каждый уровень дискретного разбавления определяют как соотношение разбавления до порогового уровня (D/T).
Смешивание атмосферного воздуха, загрязненного пахучими веществами, с отфильтрованным очищенным воздухом в дискретных объемных соотношениях достигают путем использования двух потоков воздуха:
— первого потока, проходящего через специальный фильтр;
— второго потока, проходящего через одно из отверстий в градуированном диске D/T.
Первый поток — это атмосферный воздух, очищенный от возможных примесей пахучих веществ прохождением через фильтры. Отфильтрованный воздух, не содержащий посторонних запахов, попадает внутрь ольфактометра и смешивается со вторым потоком — атмосферным воздухом с пахучими веществами, который попадает в ольфактометр из загрязненной атмосферы через одно из отверстий в градуированном диске D/T. Затем смесь отфильтрованного и пахучего воздуха подается к носу эксперта.
Разбавление до порогового уровня D/T рассчитывают по следующей формуле:
![]() , (1)
, (1)
где ![]() — объем отфильтрованного воздуха;
— объем отфильтрованного воздуха;
![]() — объем пахнущего воздуха.
— объем пахнущего воздуха.
Градуированный диск D/T позволяет осуществлять дискретное разбавление до порогового уровня, где пробу загрязненного пахучего воздуха разбавляют в 2, 4, 7, 15, 30, 60 раз и более.
Проводя исследования, эксперт плотно прикладывает прибор к носу и вдыхает разбавленную в различной степени пробу воздуха, отвечая на вопрос, чувствует запах или не чувствует.
Во втором случае используют переносной ольфактометр, который всасывает пробу загрязненного атмосферного воздуха через эжектор (насос Вентури) и разбавляет ее, используя чистый воздух, который подается из баллона. Микроконтроллер регулирует коэффициент разбавления и выводит данные на планшет с сенсорным экраном. По методу «да/нет» эксперт определяет наличие либо отсутствие запаха. Далее ольфактометр автоматически рассчитывает концентрацию запаха в единицах запаха в 1 м атмосферного воздуха.
Полевой метод позволяет оценивать наличие и концентрацию запаха в выбранных точках на местности вблизи источника выбросов запаха, например на границе санитарно-защитной зоны предприятия. Метод позволяет оценивать концентрацию, частоту и продолжительность периода запаха в окрестностях предприятия в течение длительного периода наблюдений (от 6 до 12 мес). В то же время следует отметить, что результаты, полученные полевыми исследованиями, трудно поддаются систематизации из-за нестабильности метеорологических условий, от которых зависит распространение запаха в атмосфере. Однако репрезентативность полученных результатов может быть улучшена количеством проведенных измерений: чем дольше и чаще осуществляют измерения, тем более объективную картину можно получить.
6 Инвентаризация выбросов запаха на предприятиях
Инвентаризацию выбросов запаха на предприятии проводят при наличии постоянных жалоб людей, проживающих в окрестностях предприятия, на неприятный запах, а также волеизъявления государственных органов и самого предприятия решить проблему воздействия запаха на население.
Инвентаризация выбросов запаха на предприятиях включает несколько этапов:
— органолептическое обследование промышленной площадки предприятия по ГОСТ ISO 5492, ГОСТ ISO 1600-30*;
________________
* Вероятно, ошибка оригинала. Следует читать: ГОСТ ISO 16000-30. — .
— отбор проб выбросов запаха на источнике;
— количественное ольфактометрическое измерение концентрации запаха в выбросах предварительно выбранных источников, ЕЗ/м;
— расчет мощности выбросов запаха от исследуемых источников, ЕЗ/с;
— расчет рассеивания выбросов запаха в атмосферном воздухе в окрестностях предприятия, ЕЗ/м по [2].
Результаты, полученные при инвентаризации выбросов запаха, позволяют определить вклад наиболее мощных источников выбросов запаха, на которых в первую очередь целесообразно проводить мероприятия по снижению выбросов запаха, оценить уровни воздействия запаха на население, проживающее вблизи предприятия, и подготовить необходимые материалы для обоснования и установления значения норматива запаха в атмосферном воздухе.
На первом этапе инвентаризации выбросов запаха проводят органолептическое обследование предприятия, во время которого члены экспертной группы по запахам, основываясь на собственном чувстве обоняния, выявляют источники, выбросы которых имеют ощутимый запах. Органолептическое обследование предприятия необходимо проводить с учетом технологического регламента предприятия во время наибольших выбросов пахучих веществ. Для неорганизованных источников, расположенных на открытом воздухе, предварительное обследование и инвентаризация выбросов должны быть осуществлены в летнее время.
При инвентаризации выбросов запаха от предприятий применяют метод ольфактометрического измерения концентрации запаха по 6.1 в пробах газовоздушной смеси, отобранных на источнике выбросов. Газовоздушная смесь, поступающая в атмосферу из источника выброса, может содержать одно загрязняющее вещество или представлять собой смесь веществ известного и неизвестного состава.
6.1 Измерение концентрации запаха в пробе ольфактометрическим методом
Ольфактометрический метод измерения концентрации запаха в исследуемой пробе основан на предъявлении группе отобранных в соответствии с 6.3 экспертов различных концентраций запаха, полученных путем разбавления пробы чистым воздухом, для определения фактора разбавления при достижении 50%-ного порога ощущения. Концентрация запаха, которую ощущают 50% испытуемых, считается равной 1 ЕЗ/м.
Эталонным веществом при измерении запаха является н-бутанол. Одна европейская эталонная масса запаха (EROM) эквивалентна 123 мкг н-бутанола; будучи разбавлена в 1 м воздуха, соответствует одной единице запаха (1 ЕЗ/м
). Связь между единицей запаха (ЕЗ) для эталонного пахучего вещества (н-бутанола) и ЕЗ для любой смеси пахучих веществ определяют на уровне психологической реакции (предела обнаружения) и записывают:
1 EROM = 123 мкг н-бутанола = 1 ЕЗ для смеси пахучих веществ.
Указанная связь является основой при количественном определении концентрации запаха в единицах запаха для любого пахучего вещества или смеси веществ. При ольфактометрических исследованиях концентрацию запаха измеряют путем установления фактора разведения, который необходим для достижения порога определения в 1 ЕЗ/м.
Концентрация запаха пахучего вещества, измеряемая как величина, кратная 1 ЕЗ в 1 м нейтрального газа, может быть использована при расчетах рассеивания выбросов так же, как и обычные массовые концентрации загрязняющих веществ, мг/м
.
Применяемая единица запаха связывает пахучее вещество (возбудитель) или смесь пахучих веществ с его физиологическим воздействием.
При использовании концентраций запаха необходимо помнить, что отношение между интенсивностью запаха и его концентрацией нелинейно, и могут быть разные отношения для разных одорантов (смесей). Пробы запаха анализируют в специальном помещении, которое должно соответствовать требованиям для ольфактометрической лаборатории (см. 6.2.7). Ольфактометрический анализ пробы запаха осуществляется подготовленной группой экспертов на ольфактометре. Перед началом любого измерения группа экспертов должна быть протестирована на эталонной смеси (н-бутаноле) для подтверждения их способности к работе в данный конкретный день (см. 6.3). Если эксперты ощущают посторонний запах, систему необходимо проверить и устранить причину возникновения постороннего запаха с помощью продувки ольфактометра чистым воздухом. После проверки эксперты приступают к анализу пробы.
Число экспертов в комиссии должно составлять не менее четырех человек (после исключения аномальных результатов). Каждый эксперт размещается у своего нюхательного порта (или нюхательных портов) за рабочим местом ольфактометра.
К ольфактометру присоединяют пакет (мешок) с пробой запаха, из которого отбирают пробы в систему разбавления, после чего разбавленная проба подается к нюхательным портам всех экспертов. Скорость подачи пробы должна быть комфортной с точки зрения экспертов и составлять от 0,2 до 0,5 м/с. Эксперта при проведении измерений не должны отвлекать посторонние запахи, разговоры, шумы и т.д. При подаче пробы на панели ольфактометра у каждого порта должны загораться индикаторные лампочки, что служит сигналом для эксперта. Подача пробы чередуется с подачей чистого воздуха. Если эксперт ощущает запах, он нажимает специальную кнопку, соответствующую ответу «да, запах чувствуется»; если эксперт не уверен, сомневается или четко понимает, что проба не содержит определяемого запаха, то кнопку не нажимает.
Время представления запаха не должно превышать 15 с. Время между представлениями запаха должно быть достаточным для того, чтобы избежать адаптации к запаху, причем при случайном порядке представления проб время должно составлять не менее 30 с. Интервал между подачами проб с различными степенями разбавления также должен составлять не менее 30 с.
Запах представляют испытуемым в течение одной серии разбавлений в случайном или возрастающем порядке. Разбавленные в разных соотношениях пробы подают на анализ эксперту до достижения индивидуального порога выявления запаха. Представление проб испытуемым обязательно сочетается с представлениями чистого воздуха.
После четырех раундов измерений программа ольфактометра, установленная на подключенном к прибору компьютере, рассчитывает измеренную концентрацию.
Серии измерений, используемые в обработке результатов измерений, должны отвечать следующим критериям:
— измерения должны выявить индивидуальные пороговые оценки;
— каждая серия должна содержать не менее двух последовательных правильных ответов;
— из расчета исключаются испытуемые, положительные ответы которых на чистый воздух составили более 20%.
В одной серии измерений участвуют четыре эксперта по запахам или более, размещающиеся вокруг прибора, каждый эксперт — около одного из нюхательных портов. Эксперту предлагают оценить подаваемый из порта образец и ответить, ощущает ли он запах (да/нет). При этом испытуемым известно, что в некоторых случаях им может быть предъявлен чистый воздух.
Описание метода принудительного выбора представлено в 5.1.
6.2 Оборудование и материалы, используемые в ольфактометрии
6.2.1 Аппаратура для разбавления проб (ольфактометр)
Ольфактометр — устройство, в котором пахучее вещество, разбавленное чистым воздухом в разных пропорциях (как правило, диапазон разбавления от 2 до 2
![]() ) и в разных концентрациях, подается через нюхательный порт членам экспертной группы для оценки. Измерения могут проходить в порядке увеличения концентрации, начиная с концентраций ниже порога восприятия (предельный метод), или в случайной последовательности — выше и ниже порога восприятия (непрерывный метод). Эксперт должен выбрать один из ответов — «да, пахнет» или «нет, не пахнет». С помощью ольфактометра, понижая степень разбавления вещества воздухом, измеряют порог восприятия (одну единицу запаха в 1 м
) и в разных концентрациях, подается через нюхательный порт членам экспертной группы для оценки. Измерения могут проходить в порядке увеличения концентрации, начиная с концентраций ниже порога восприятия (предельный метод), или в случайной последовательности — выше и ниже порога восприятия (непрерывный метод). Эксперт должен выбрать один из ответов — «да, пахнет» или «нет, не пахнет». С помощью ольфактометра, понижая степень разбавления вещества воздухом, измеряют порог восприятия (одну единицу запаха в 1 м), концентрацию запаха, которую способны воспринимать 50% экспертов. Измерение запаха на ольфактометре позволяет получить его концентрацию в единицах запаха, ЕЗ/м
.
Всю программу разбавления контролируют на компьютере и выполняют автоматически. Программа разбавления включает несколько последовательных измерений, восстановительные периоды для испытуемых и продувку блока разбавления чистым воздухом.
Должны быть минимизированы время воздействия, длина и диаметр газопроводящей системы ольфактометра для подачи и предъявления проб запаха членам экспертной группы, а также для предотвращения их загрязнения запахом. Необходимо избегать устройств, способных повлиять на свойства газов или проб.
Температура предъявляемого испытуемым нейтрального газа или пробы не должна отличаться более чем на 3°С от комнатной.
Ольфактометр должен обеспечивать отношение максимального разбавления к минимальному не менее 2.
Порты ольфактометра для предъявления проб экспертам должны отвечать следующим требованиям:
— устройство должно позволять испытуемому вдыхать и выдыхать без затруднений;
— поток воздуха из порта должен составлять не менее 20 л/мин.
6.2.2 Требования к материалам, используемым в ольфактометрии
Требования к материалам, используемым для определения запаха методом ольфактометрии:
— не должны иметь запаха;
— должно быть сведено к минимуму физическое или химическое взаимодействие между применяемыми материалами и исследуемыми пробами;
— не должны вызывать потерю пробы вследствие диффузии;
— должны иметь гладкую поверхность.
Материалы, используемые и соприкасающиеся с исследуемыми пробами в оборудовании: политетрафторэтилен, сополимер тетрафторэтилена и гексафторпропилена, полиэтилентерефталат (налофан), стекло и др. Не допускается использовать силикон и резину.
Использованные материалы перед применением необходимо тщательно очистить от следов пахучих веществ.
Материалы, используемые для контейнеров (пакетов) и отбора проб: сополимер тетрафторэтилена и гексафторпропилена, полиэтилентерефталат (налофан), поливинилфторид (тедлар). Материалы для пакетов перед применением должны быть проверены на наличие посторонних запахов.
6.2.3 Вакуумное устройство для отбора проб запаха
Данное устройство предназначено для отбора проб запаха, состоит из вакуумируемого сосуда, в котором вакуум создается вакуумным насосом с питанием от аккумуляторной батареи. Вакуумный насос и батареи расположены в нижней части устройства. Пластиковый прозрачный корпус вакуумного устройства позволяет наблюдать за процессом наполнения пакета (мешка). Время наполнения пакета (мешка) для отбора проб должно составлять 20 мин.
Вакуумное устройство используют для отбора газообразных проб. Его необходимо предохранять от физического воздействия и воздействия чрезмерно высоких температур.
6.2.4 Пакеты (мешки) для отбора проб
Пробы запаха отбирают в пакеты (мешки) с диаметром 150 мм. Для формирования нижней части пакета (мешка) один свободный конец заготовки пакета (мешка) складывают «гармошкой» длиной складки около 10 мм, свернутый конец загибают для получения двойного слоя, составляющего около 30 мм. Для герметизации край пакета (мешка) жестко фиксируют одним или двумя кабельными хомутами (стяжкой), лишнюю часть хомута отрезают.
Для формирования верхней части пакета (мешка) конец пакета (мешка) также складывают «гармошкой» примерно до середины пакета (мешка) по ширине с длиной складки примерно 10 мм. В середину пакета (мешка) помещают трубку длиной от 15 до 16 см, внутренний диаметр которой составляет 6 мм, внешний диаметр — 8 мм, изготовленную из нержавеющей стали или политетрафторэтилена. Конец трубки, помещенный внутрь пакета (мешка), не должен быть зажат между складками, а должен находиться в свободном состоянии для беспрепятственного проникновения воздуха в пакет (мешок), что необходимо для отбора пробы и ее дальнейшего анализа на ольфактометре. Другую часть пакета (мешка) собирают в «гармошку» и закрепляют, как и в случае нижнего конца пакета (мешка), одним или двумя кабельными хомутами. Лишнюю часть хомута отрезают.
Трубку затыкают корковой пробкой, диаметр нижнего конца которой составляет около 4 мм, а диаметр другого конца не должен превышать 7 мм, при этом длина пробки — около 16 мм.
6.2.5 Очистка оборудования для его повторного использования
Оборудование для отбора проб должно быть очищено от любых пахучих веществ. После использования трубки следует тщательно промыть водой в случае сильного загрязнения, когда трубки были подвергнуты воздействию высоких концентраций запаха, трубки промывают мыльным раствором, который не должен иметь сильного запаха парфюмерных отдушек. После промывки водой трубки высушивают сжатым воздухом. Хранят трубки в герметичном полиэтиленовом пакете.
Пакеты (мешки) для отбора проб запаха, изготовленные, как правило, из полиэтилентерефталата (налофан), должны быть протестированы для определения фоновой концентрации запаха и проверены на отсутствие утечек.
Тест для определения фоновой концентрации запаха материала для мешка следует проводить путем заполнения нейтральным газом не менее трех мешков, сделанных из тестируемого материала, и хранения их в течение 30 ч. Затем выполняют измерение запаха в каждом мешке для определения концентрации. Можно считать, что материал для мешка не имеет запаха, если порог запаха не был достигнут во всех мешках.
Мешки не могут быть повторно использованы до тех пор, пока каждый мешок не проверят на отсутствие запахов в соответствии с вышеописанной процедурой.
6.2.6 Требования к газам, используемым в ольфактометрии
Воздух или нейтральный газ, предназначенный для разведения проб, должен быть безопасен для вдыхания экспертами и не должен обладать запахом. Для разведения проб можно использовать следующее: сжатый воздух из компрессора (рекомендуется безмасляный компрессор), прошедший фильтрацию, охлаждение, высушивание и очищение активным углем; азот из баллонов с жидким азотом; окружающий воздух из хорошо кондиционированной комнаты; синтетический воздух из баллонов.
Используемый в качестве эталона н-бутанол должен быть высокой очистки.
6.2.7 Требования к помещению для ольфактометрических измерений по ГОСТ ISO 8589, ГОСТ Р 53701
Помещение лаборатории, в котором проводят ольфактометрические измерения:
— должно быть хорошо проветриваемым, оснащенным вентиляцией, соответствовать общим санитарным нормам;
— в воздухе должен отсутствовать запах;
— не допускаются резкие перепады температур, максимальная температура не должна превышать 25°С, колебание температуры во время проведения исследований не должно превышать ±3°С;
— не должно подвергаться воздействию прямых солнечных лучей, световому и шумовому воздействию;
— не допускается выделение пахучих веществ от оборудования, мебели, покрытий пола, стен и др.
6.3 Формирование экспертной группы для измерения концентрации запаха
В состав экспертной группы для измерения концентрации запаха в отобранных пробах включают добровольцев, которые после прохождения серии испытаний соответствуют всем критериям отбора. Основным критерием выбора экспертов для измерения концентрации запаха является их индивидуальная чувствительность к запахам.
Результатом процедуры отбора проб является показатель ITE (индивидуальное пороговое значение), который рассчитывают на основе не менее десяти исследований по измерению пороговой оценки эталонного вещества. В качестве эталонного вещества используют газовую смесь н-бутанола в азоте (воздухе).
Каждый эксперт участвует в трех сериях испытаний, каждую из которых проводят в отдельный день, между сериями должно быть не менее дня. Результаты, полученные в ходе этих испытаний, должны соответствовать следующим критериям:
— антилогарифм стандартного отклонения ![]() , рассчитанный из десятичных логарифмов индивидуальных пороговых оценок ITE, выраженный в единицах массовой концентрации эталонного газа (н-бутанола), не должен превышать 2,3;
, рассчитанный из десятичных логарифмов индивидуальных пороговых оценок ITE, выраженный в единицах массовой концентрации эталонного газа (н-бутанола), не должен превышать 2,3;
— среднее геометрическое значение индивидуальных пороговых оценок ITE, выраженное в единицах массовой концентрации эталонного газа, должно попасть в диапазон 62-246 мкг/м для н-бутанола.
При измерении концентрации запаха ольфактометрическим методом экспертную группу из четырех человек или более размещают за прибором, каждый около своего индивидуального порта (одновременно или поочередно). Оператор подсоединяет пакет с эталонной смесью н-бутанола, включает программу, и ольфактометр начинает подавать, разбавленный чистым воздухом эталонный газ из пакета (мешка) для отбора проб в каждый нюхательный порт. Кратность разбавления, как правило, составляет от 2 до 2
![]() . Эксперт, нажимая или не нажимая на специальную кнопку, должен отметить, чувствует ли он запах в тот момент, когда подача разбавленной пробы выпадает на его порт, или не чувствует. По окончании измерения программа автоматически рассчитывает результат и выводит его на экран монитора. Результаты сохраняют в компьютере и могут быть распечатаны.
. Эксперт, нажимая или не нажимая на специальную кнопку, должен отметить, чувствует ли он запах в тот момент, когда подача разбавленной пробы выпадает на его порт, или не чувствует. По окончании измерения программа автоматически рассчитывает результат и выводит его на экран монитора. Результаты сохраняют в компьютере и могут быть распечатаны.
Помимо прочего отбор экспертов для ольфактометрии может быть осуществлен по методикам производителей ольфактометров, например с помощью специальных маркеров.
В измерениях не могут принимать участие лица с простудными заболеваниями, аллергией, заболеваниями носовых пазух и т.п. Для возможности адаптации членов экспертной группы впускают в рабочее помещение за 15 мин до начала проведения измерений.
Члены экспертной группы до проведения ольфактометрического измерения должны соблюдать следующие требования:
— за 30 мин до проведения измерения не должны курить, есть, пить (за исключением воды), использовать жевательную резинку или сладости;
— должны внимательно следить за личной гигиеной и быть осторожными в использовании парфюмерных средств, дезодорантов, лосьонов для тела, духов и т.п.;
— во время проведения измерений не должны общаться между собой и обмениваться мнениями.
6.4 Отбор проб
Пробы запаха отбирают в пакет (мешок) по 6.2.4. Пакет (мешок) устанавливают в оборудование для отбора проб запаха. Рекомендуется перед отбором пробы пакет (мешок) несколько раз заполнить пробой запаха и спустить ее. Заполнение пакета (мешка) происходит непрерывно или прерывисто, путем короткого включения и выключения оборудования для отбора проб, равномерно по всему объему.
Во время отбора пробы может образоваться конденсат, проба газовоздушной смеси может быть слишком горячей, содержать примеси, например пыль, или быть чрезвычайно пахучей. Для таких ситуаций должны быть предусмотрены специальные процедуры. При высоких концентрациях запаха, превышающих верхний предел определения ольфактометра, следует предварительно разбавить пробу в необходимом соотношении методом динамического разбавления с помощью устройства предварительного разбавления. При отборе пылегазовоздушной смеси для предотвращения попадания пыли в пакет (мешок) используют обычные фильтры.
При отборе пробы запаха анализируемый воздух или газовоздушная смесь не должны перемешиваться с другими различными потоками. Для проведения ольфактометрического анализа необходимо отбирать достаточное количество проб (не менее трех). На каждый пакет (мешок) с пробой запаха приклеивают этикетку, на которой указывают дату и время отбора пробы, источник отбора, температуру воздуха и атмосферное давление.
Анализ проб запаха необходимо проводить, по возможности, непосредственно после отбора. Интервал между отбором проб и проведением измерений не должен превышать 30 ч, так как при хранении в пробе могут происходить изменения, влияющие на первоначальный запах. Со временем возрастает вероятность протекания процессов адсорбции, диффузии, химического взаимодействия, что может привести к изменению состава отобранных проб. Во время транспортирования и хранения проб температура не должна превышать 25°С. Во избежание конденсации температура не должна опускаться ниже точки росы.
По 4.2.1 ГОСТ 17.2.3.02-2014 продолжительность отбора проб запаха так же, как и загрязняющих веществ, должна составлять 20 мин. Если время отбора одной пробы запаха менее 20 мин, то за 20-минутный период необходимо отобрать не менее трех проб, а результаты осреднить.
В выбросах при высоком содержании пыли или влаги перед устройством для отбора пробы устанавливают фильтр или каплеотбойник.
Для определения концентрации запаха и дальнейшего расчета мощности выбросов запаха одновременно с отбором проб осуществляют измерение физических параметров газовых потоков. К измеряемым параметрам газовых потоков относят: температуру по ГОСТ 17.2.4.07, давление (разрежение) по ГОСТ 17.2.4.07, влажность по ГОСТ 17.2.4.08 и скорость газа в газоходе по ГОСТ 17.2.4.06. Применяемая аппаратура и оборудование для измерения физических параметров газовых потоков промышленных выбросов должны быть аттестованы, внесены в Государственный реестр средств измерений и иметь методическое обеспечение.
Температуру измеряют лабораторными термометрами, цифровыми термометрами или термопарами. Основными критериями выбора средства измерений являются диапазон измеряемых температур и стойкость средства измерения к газовой среде. Измерения температуры проводят не менее трех раз и рассчитывают среднее значение. Погрешность измерения температуры оценивают по ГОСТ Р 8.736.
Давление в газоходе определяют с помощью пневмометрических трубок, подсоединенных к манометрам. Различные виды пневмометрических трубок и манометры выбирают в зависимости от параметров газового потока. При этом температура газа в газоходе не должна превышать 400°С, а скорость газового потока должна быть от 4 до 10 м/с. Способы измерения скорости с применением пневмометрических трубок оценивают по ГОСТ 17.2.4.06.
Неорганизованный источник выбросов запаха представляет собой открытую поверхность (пруды-отстойники, накопители, свалки и т.д.). Количественное определение концентраций и выбросов загрязняющих веществ, включая запах от такого рода источников, — очень сложная задача. В случае неорганизованных источников пробы запаха отбирают в виде загрязненного пахучими веществами атмосферного воздуха, при этом отбор проб осуществляют в нескольких точках в непосредственной близости от источника с наветренной и подветренной сторон. Для каждого конкретного неорганизованного источника составляют свою программу отбора проб с указанием, в частности, точек отбора, с учетом расположения источника, скорости и направления ветра.
Кроме того, для отбора проб воздуха от неорганизованных источников можно применять специальные пробоотборные колпаки. Для аэрируемых источников с невысокой скоростью потока воздуха используют невентилируемый колпак, который перекрывает источник и канализирует поток воздуха в трубе, в отверстие которой помещают пробоотборное устройство. Для неаэрируемых источников применяют вентилируемый колпак, в котором с помощью двух вентиляторов создается постоянный расход воздуха. Наружный воздух всасывается с помощью приточного вентилятора, очищается на угольном фильтре, а затем распределяется по всему объему колпака и абсорбирует пахнущие вещества. После этого с помощью другого вентилятора воздух вытягивается к противоположному краю системы, откуда через всасывающие сопла попадает внутрь пробоотборного устройства, закрепленного на колпаке.
Число отобранных проб должно быть достаточным для проведения ольфактометрического анализа с учетом их разбавления и составлять не менее трех.
6.5 Расчет выбросов запаха
Величину мощности выброса запаха, ЕЗ/с, на конкретном источнике определяют на основе ольфактометрических измерений концентрации запаха в выбросе и объемного расхода газовоздушной смеси.
Разовое значение мощности выброса запаха ![]() , ЕЗ/с, для организованного источника для каждой пробы без учета влияния давления рассчитывают по формуле
, ЕЗ/с, для организованного источника для каждой пробы без учета влияния давления рассчитывают по формуле
![]() , (2)
, (2)
где ![]() — концентрация запаха в k-пробе, измеренная в лабораторных условиях (20°С и 101,3 кПа), ЕЗ/м
— концентрация запаха в k-пробе, измеренная в лабораторных условиях (20°С и 101,3 кПа), ЕЗ/м;
![]() — полный объем газовоздушной смеси, измеренный в процессе отбора k-пробы, м
— полный объем газовоздушной смеси, измеренный в процессе отбора k-пробы, м/с (включая объем водяных паров), выбрасываемой в атмосферу из устья источника в течение 1 с при температуре газовоздушной смеси
![]() ;
;
![]() — стандартная температура в ольфактометрии — 20°С;
— стандартная температура в ольфактометрии — 20°С;
![]() — температура газовоздушной смеси на выходе из источника, °С.
— температура газовоздушной смеси на выходе из источника, °С.
Фактическую мощность выброса запаха на конкретном источнике g, ЕЗ/с, рассчитывают как среднее геометрическое значение величин выбросов для отобранных проб (например, для трех проб) по формуле
![]() . (3)
. (3)
7 Расчет рассеивания и нормирование выбросов запаха
Запах, исходящий от источников выбросов, связан с поступлением в атмосферу разнообразных пахучих веществ, совокупные выбросы которых и создают неприятные для человека ощущения. Закономерности переноса индивидуального пахучего соединения и смеси веществ, определяющих запах в атмосферном воздухе, одинаковы. Для изучения распространения запаха в атмосфере применяют те же математические модели, что и для расчета рассеивания выбросов загрязняющих веществ. В настоящее время в Российской Федерации используют Методы расчетов рассеивания [2]. Процедура установления нормативов выбросов запаха на конкретных источниках полностью совпадает с аналогичной процедурой в случае выбросов индивидуальных загрязняющих веществ. Рассчитанные значения концентрации запаха, формируемые его выбросами на границе жилой зоны, сравнивают с нормативной концентрацией запаха в атмосферном воздухе. При отсутствии превышения норматива запаха в атмосферном воздухе мощность выброса запаха, ЕЗ/с, определенная при инвентаризации, рассматривается как норматив предельно допустимого выброса запаха для данного источника.
8 Контроль за соблюдением нормативов запаха
Контроль установленных нормативов выбросов запаха должен быть осуществлен на источнике выбросов запаха или на основе натурных исследований запаха.
Контроль на источнике выбросов запаха включает:
— отбор проб выбросов запаха на источнике контролируемого предприятия;
— ольфактометрический анализ отобранных проб для определения концентрации запаха в выбросах, ЕЗ/м;
— расчет мощности выбросов запаха, ЕЗ/с;
— расчет рассеивания выбросов запаха в окрестностях предприятия.
При сравнении рассчитанной максимальной концентрации запаха на границе жилой зоны с установленным нормативным значением запаха в атмосферном воздухе делается вывод о соблюдении или несоблюдении норматива запаха. Указанный способ является достаточно трудоемким и дорогостоящим. Он применяется при первоначальном исследовании выбросов запаха на предприятии, на основе результатов которого устанавливают нормативное значение концентрации данного запаха в атмосферном воздухе конкретного населенного пункта. Повторные измерения выбросов запаха с последующим расчетом рассеивания проводят при изменении сырья, технологического процесса, модернизации производства, применении мероприятий по снижению выбросов запаха и т.д., а также по предписанию контролирующих органов.
При контроле за соблюдением норматива запаха на основе натурных исследований концентрацию запаха измеряют специально обученные инспекторы с помощью переносных ольфактометров непосредственно в воздухе населенных мест, прилегающих к предприятию.
Программу натурных исследований разрабатывают для конкретного случая с учетом климатических и географических условий, рельефа местности, периодичности выбросов запаха, видов источников запаха и др. Для количественной оценки загрязненности запахом атмосферного воздуха можно использовать полевую ольфактометрию. Переносные ольфактометры (см. 5.2.2) являются экономически эффективным средством количественной оценки запаха в виде соотношения D/T или ЕЗ/м. Переносной ольфактометр для мониторинга запаха в атмосферном воздухе в определенных точках на территории объекта или непосредственно в жилой зоне могут использовать сотрудники предприятия, на котором происходит выброс пахучих веществ, инспекторы контролирующих органов и население, проживающее на прилегающей к предприятию территории, только после соответствующей подготовки в соответствии с требованиями настоящего стандарта. Переносные ольфактометры рекомендуют применять для следующих видов мониторинга запаха в атмосферном воздухе:
— мониторинг на объекте — осуществление мониторинга запахов в различных предварительно выбранных точках (например, открытых дверных проемах, въездах, складах и по периметру ограждения) на территории предприятия на протяжении определенного периода времени (дня, недели и т.д.). Мониторинг может включать также исследование (на предмет наличия запаха) используемых на предприятии материалов, производственной деятельности вне помещений и неорганизованных атмосферных выбросов;
— произвольный мониторинг, при применении которого используют подход «выборочного инспектирования». Произвольный мониторинг позволяет собрать данные, которые могут быть соотнесены с метеорологической информацией и деятельностью, осуществляемой на объекте;
— запланированный мониторинг — заранее спланированный мониторинг может состоять из ежедневного (ежеквартального, годового) «обхода» или «объезда» (или запланированных нескольких визитов) предварительно обозначенных точек мониторинга. Данные, полученные с помощью полевого ольфактометра, могут быть использованы для коррекции различных параметров, которые оказывают воздействие на возникновение запаха (например, производственную деятельность на объекте);
— исследование запахов на организованных и неорганизованных источниках выбросов запаха позволяет определить, какие источники или деятельность могут вызывать запах за пределами объекта, а какие не могут. Все потенциальные источники запахов и соответствующие операции могут быть классифицированы в соответствии с тем вкладом, который они вносят в образование запахов;
— мониторинг в жилой зоне — применение мониторинга может стать частью интерактивной программы работы с населением. Основной задачей данного вида мониторинга является сбор информации посредством точного ведения учета реальных условий в жилой зоне. Собрать информацию о характеристиках запахов могут помочь граждане, специально обученные методам измерения запахов с помощью полевого ольфактометра. Осуществление мониторинга в жилой зоне способствует определению периодов времени и погодных условий, которые характерны для образования запахов. Мониторинг в жилой зоне с помощью полевого ольфактометра помогает определить интенсивность запаха, при которой запах начинает оказывать неблагоприятное воздействие;
— реакция на поступающие жалобы — «горячие линии», принимающие жалобы по поводу наличия неприятных запахов, могут стать удобной практикой, используемой предприятиями и соответствующими контролирующими органами для своевременной реакции на факты возникновения запахов. План мероприятий реагирования на поступившие жалобы (включающий конкретных лиц, ответственных за оперативное проведение соответствующих действий) дает возможность установить возникновение запаха, обнаружить источник запаха и количественно оценить интенсивность запаха;
— составление профиля шлейфа запахов — моделирование рассеивания запаха в атмосферном воздухе позволяет составлять прогнозы распространения запаха вокруг предприятия. Составление профиля шлейфа запахов инструментальным методом дополняет и «калибрует» результаты моделирования рассеивания в атмосферном воздухе. Несколько инспекторов с полевыми ольфактометрами располагаются против и по направлению ветра от источника запаха, измеряют и фиксируют интенсивность запаха в виде соотношений D/T или ЕЗ/м. Профиль шлейфа запаха затем документируют и накладывают на карту местности. Моделирование рассеивания в атмосферном воздухе и топография местности могут быть соотнесены с фактическими измерениями запахов, осуществляемыми с помощью полевой ольфактометрии.
9 Мониторинг интенсивности запаха
Альтернативный метод контроля качества атмосферного воздуха — это мониторинг интенсивности запаха.
При контроле качества атмосферного воздуха на основе натурных исследований мониторинг воздействия запаха осуществляют специально обученные инспекторы, оценивающие интенсивность запаха органолептическим способом непосредственно в воздухе населенных мест, прилегающих к предприятию, согласно ГОСТ ISO 16000-30. Оценку интенсивности запаха проводят по 5-балльной шкале в соответствии с таблицей 2.
Таблица 2 — Интенсивность и характер запаха
|
Интенсивность запаха, балл |
Характеристика интенсивности запаха |
Описание проявления запаха |
|
0 |
Запах отсутствует |
Отсутствие ощутимого запаха |
|
1 |
Очень слабый |
Запах, как правило, не замечаемый, но обнаруживаемый инспектором, если он специально обращает на этот запах внимание |
|
2 |
Слабый |
Слабый запах, обнаруживаемый инспектором, но еще не вызывающий негативной реакции |
|
3 |
Отчетливый |
Заметный запах, который может вызвать негативную реакцию у инспектора |
|
4 |
Сильный |
Запах, обращающий на себя внимание и вызывающий негативную реакцию у инспектора |
|
5 |
Очень сильный |
Запах, настолько сильный, что вызывает очень неприятные ощущения у инспектора |
Библиография
|
[1] |
EH 13725:2003 Качество воздуха. Определение концентраций запахов методом динамической ольфактометрии |
|
[2] |
Методы расчетов рассеивания выбросов вредных (загрязняющих) веществ в атмосферном воздухе (МРР-2017) |
|
УДК 504.3.054:006.354 |
ОКС 01.040.13 |
|
Ключевые слова: запах, нормативы, выбросы, атмосферный воздух |
Электронный текст документа
и сверен по:
, 2019
УТВЕРЖДАЮ
Заместитель Главного
санитарного врача СССР
Д.Н.ЛОРАНСКИЙ
2 февраля 1971 г. N 880-71
ИНСТРУКЦИЯ
ПО САНИТАРНО-ХИМИЧЕСКОМУ ИССЛЕДОВАНИЮ ИЗДЕЛИЙ, ИЗГОТОВЛЕННЫХ ИЗ ПОЛИМЕРНЫХ И ДРУГИХ СИНТЕТИЧЕСКИХ МАТЕРИАЛОВ, ПРЕДНАЗНАЧЕННЫХ ДЛЯ КОНТАКТА С ПИЩЕВЫМИ ПРОДУКТАМИ
(в ред. Методических указаний, утв. заместителем Главного государственного санитарного врача СССР 30.06.87 N 4395-87)
Настоящая Инструкция предназначена для гигиенических институтов и лабораторий санитарно-эпидемиологических станций, а также производственных институтов и лабораторий, осуществляющих повседневный контроль за соответствием посуды, тары и других изделий гигиеническим требованиям.
Инструкция подготовлена специалистами Московского научно-исследовательского института гигиены им. Ф.Ф. Эрисмана и Отделом гигиены питания Главного санитарно-эпидемиологического управления Минздрава СССР.
В работе по подготовке Инструкции принимали участие представители Всесоюзного научно-исследовательского института гигиены и токсикологии пестицидов, полимерных и пластических масс; токсикологической лаборатории научно-производственного объединения «Пластполимер»; проблемной лаборатории и кафедры полимеров Московского технологического института мясной и молочной промышленности; лаборатории новых материалов Всесоюзного научно-исследовательского молочного института и др.
В обсуждении проекта Инструкции активное участие принимали представители Института питания АМН СССР, Института гигиены водного транспорта, Всесоюзного научно-исследовательского института консервной и овощесушильной промышленности, Всесоюзного научно-исследовательского института мясной промышленности, Всесоюзного научно-исследовательского и экспериментально-конструкторского института продовольственного машиностроения, санитарно-эпидемиологические станции г. Москвы и Московской области, г. Ленинграда и Харьковской области.
С изданием настоящей Инструкции «Инструкция по санитарно-химическому испытанию новых видов пищевой посуды, тары и других изделий, изготовленных с применением синтетических лаков, эмалей, клея, резины, шпаклевки и пластмасс» (1964) утратила силу.
ВВЕДЕНИЕ
Применение синтетических полимерных материалов в различных отраслях народного хозяйства, в том числе производящих продукты питания, позволяет механизировать и автоматизировать производство, способствует развитию пищевой промышленности, упаковочной техники, повышению культуры торговли.
Благодаря высокой технико-экономической эффективности ассортимент и тоннаж полимерных материалов, внедряемых в различные отрасли пищевой промышленности, растет с каждым днем.
Вместе с тем особенности строения и свойств этих материалов обусловливают возможность перехода из них в окружающую среду химических веществ, способных в ряде случаев оказывать отрицательное влияние на качество пищевых продуктов и здоровье людей.
В связи с изложенным, с целью предупреждения возможного неблагоприятного влияния изделий из полимерных материалов на качество пищевых продуктов и здоровье людей, все контактирующие с пищевыми продуктами изделия, изготовленные с применением полимерных и других синтетических материалов, подлежат обязательной гигиенической регламентации, основанной на результатах специальных санитарно-химических и токсикологических исследований.
Наряду с этим, поскольку качество изделий из полимерных материалов в гигиеническом отношении может изменяться в процессе переработки последних в зависимости от разных условий, в ОТК предприятий должна проводиться проверка качества партий выпускаемой продукции и соответствия ее показателей определенным гигиеническим регламентам.
Гигиенические требования, условия и методы проведения указанных исследований излагаются в настоящей Инструкции.
ГИГИЕНИЧЕСКИЕ ТРЕБОВАНИЯ К ИЗДЕЛИЯМ ИЗ ПОЛИМЕРНЫХ МАТЕРИАЛОВ (ПОСУДЕ, ТАРЕ, УПАКОВКЕ И Т.П.), ПРЕДНАЗНАЧЕННЫХ ДЛЯ КОНТАКТА С ПИЩЕВЫМИ ПРОДУКТАМИ
1. Исследуемый образец не должен отдавать в воздушную среду и в контактирующие с ним модельные растворы вещества в количествах, вредных для здоровья человека, превышающих допустимые количества миграции, а также соединения, способные вызывать канцерогенный, мутагенный и другие отдаленные эффекты.
2. Поверхность образца должна быть чистой, гладкой, без раковин, трещин, наплывов, неровностей и не липкой. Внутренняя поверхность образца должна иметь светлый тон. Образец не должен иметь запаха выше одного балла.
В случае наличия одного из вышеперечисленных дефектов образец без дальнейших исследований признается непригодным для использования по назначению.
3. Внешний вид образца не должен изменяться при воздействии на него соответствующих модельных растворов, имитирующих пищевые продукты, а также при контакте с пищевыми продуктами в процессе опытной эксплуатации.
4. Исследуемый образец не должен изменять органолептических свойств модельных растворов, имитирующих пищевые продукты, после контакта с ними при соответствующих условиях.
При наличии в модельных растворах, контактировавших с образцом, одного из нижеперечисленных изменений: запаха выше 1 балла, постороннего вкуса, изменения прозрачности и цвета растворов — образец без дальнейших исследований признается непригодным для контакта с пищевыми продуктами.
Органолептические свойства пищевых продуктов после контакта их с исследуемым образцом в процессе опытной эксплуатации не должны изменяться, т.е. пищевые продукты не должны иметь каких-либо особенностей по сравнению с контрольными пищевыми продуктами.
5. Исследованные образцы, признанные удовлетворительными на основании данных санитарно-химических исследований, могут допускаться к использованию либо, в случае необходимости, подвергаться дальнейшему испытанию в условиях опытной эксплуатации. При этом обязательно участие представителя органов санэпидслужбы, установление сроков и порядка опытной эксплуатации.
По истечении установленного срока опытной эксплуатации изделий заинтересованные организации комиссионно, с участием представителя органов санэпидслужбы, проводят органолептические исследования пищевых продуктов, контактировавших с исследуемыми изделиями, отмечают состояние внутренней поверхности образца и составляют акт о результатах опытной эксплуатации.
6. На основании положительных данных, полученных при санитарно-химическом исследовании образца, а также в условиях опытной эксплуатации, органами санэпидслужбы выдается разрешение на использование изделий в условиях, в которых производилась опытная эксплуатация.
7. На изделиях, разрешенных для использования в пищевой промышленности и быту, должно быть указано: а) название материала, из которого изготовлено изделие, и марка его; б) название завода-изготовителя и область применения (товарный знак).
8. К изделию должен быть приложен проспект, в котором должно быть указано: а) для каких целей и для контакта с какими продуктами предназначается данное изделие; б) условия его использования; в) способы мойки.
9. Каждая партия изделий должна иметь сертификат, в котором должны быть указаны: завод-изготовитель, дата выпуска, номер партии, наименование материала, ГОСТ, ТУ, МРТУ на материал и изделие, дата проведения исследования в заводской лаборатории, фамилия ответственного за проверку, разрешение органов здравоохранения.
10. Все ранее разрешенные синтетические материалы, предназначенные для изготовления изделий или для покрытия металлических емкостей с целью защиты их от коррозий и других целей, не вошедшие в перечень материалов, разрешенных Главным санитарно-эпидемиологическим управлением Министерства здравоохранения СССР и союзных республик, прилагаемый к данной Инструкции, могут использоваться по назначению не более 2 лет с момента вступления в силу настоящей Инструкции. На дальнейшее использование этих материалов заинтересованные организации должны получить соответствующие разрешения.
ПОРЯДОК НАПРАВЛЕНИЯ И ПРАВИЛА ПРИЕМА ОБРАЗЦОВ ДЛЯ ЛАБОРАТОРНОГО ИССЛЕДОВАНИЯ
Новые синтетические материалы, как отечественные, так и импортные, для изготовления оборудования, посуды, тары и упаковки, предназначенные для использования в пищевой промышленности и быту, могут быть допущены только с разрешения Главного санитарно-эпидемиологического управления Министерства здравоохранения СССР.
В связи с этим заинтересованные организации должны представить образцы и необходимые материалы в Главное санэпидуправление Министерства здравоохранения СССР, которое, в случае необходимости, может направить их для экспертного заключения в соответствующие институты или лаборатории системы Министерства здравоохранения.
Образцы или модели посуды, тары, упаковочного материала и других изделий могут быть приняты для исследования институтом или лабораторией системы Министерства здравоохранения только при наличии поручения органов санитарно-эпидемиологической службы.
Образцы для исследования представляются в натуральную величину, если они не громоздки, в противном случае представляются модели их емкостью не более 1 литра. В тех случаях, когда лаками, шпаклевками и т.п. покрывается или обрабатывается не вся поверхность изделия, а только отдельные ее участки (как, например, клепка с сучками, трещинами и пр.), для лабораторного исследования могут быть представлены пластинки размером 4 x 5 см, покрытые синтетическими материалами со всех сторон, включая и торцы, по той же технологии, которая разработана для покрытия изделия.
Образцы изделий, предназначенные для исследования, должны быть изготовлены по той же технологии, которая будет применяться при массовом производстве данных изделий, и представлены для исследования не менее чем через 10 дней после их изготовления.
Изготовление образцов должно быть качественным, без дефектов. Поверхность должна быть чистой, гладкой, без раковин, трещин, наплывов, неровностей, не липкой. Образец не должен иметь запаха выше 1 балла. Внутренняя поверхность образца должна быть светлого тона.
При наличии одного из дефектов образец не может быть принят для лабораторного исследования.
Количество образцов, необходимых для испытания, зависит от характера и объема исследования и согласовывается заинтересованной организацией с учреждением, которое будет проводить исследование. Минимальное количество — 5 экземпляров. Количество упаковочного материала должно быть около 1 кв. м.
Одновременно с образцами заинтересованные организации представляют следующие сведения:
1. Наименование материала, из которого изготовлено изделие (модель), марка его и ГОСТ или ТУ на данный материал и готовое изделие.
2. Рецептуру материала, использованную для изготовления представленных для исследования образцов (моделей). Наряду с торговыми названиями веществ, входящих в рецептуру, должно быть дано их полное химическое название; должны быть указаны остаточные количества мономера и других веществ в материале и готовом изделии, теплостойкость материала.
3. Краткое описание технологии изготовления материала и образцов (моделей) с указанием температурного режима их изготовления.
4. Сведения о том, с какими пищевыми продуктами будет контактировать изделие и соотношение площади поверхности изделия к весу или объему пищевого продукта.
5. Условия эксплуатации изделия (время контакта, температурный режим и т.д.).
6. Результаты испытаний, выполненных производственными лабораториями по настоящей Инструкции, а также отчеты об опытах, если таковые проводились заинтересованной организацией.
7. В тех случаях, когда в состав рецептуры входят малоизвестные ингредиенты и в инструкции нет методов их определения, ведомственные институты (лаборатории) должны разработать чувствительные микрометоды их определения в соответствующих растворах и вместе с вышеуказанными материалами представить в Главное санитарно-эпидемиологическое управление Министерства здравоохранения СССР. Наряду с этим для апробации методов определения должны быть доставлены малоизвестные ингредиенты в чистом виде.
8. Если изделие изготовлено из импортных синтетических материалов, необходимо представить соответствующий документ о качественном составе материала и подтверждающий, что данный материал допущен в экспортирующей стране для изготовления изделий для пищевой промышленности (указав, для контакта с какими пищевыми продуктами они предназначены), а также заключение о его безвредности и результаты исследований.
9. Сведения о том, какие моющие средства будут применяться при использовании изделий на практике.
Примечание. Образцы, представленные для исследования, обратно заказчику не возвращаются.
СХЕМА САНИТАРНО-ХИМИЧЕСКОГО ИССЛЕДОВАНИЯ ИЗДЕЛИЙ ИЗ ПЛАСТМАСС И ДРУГИХ СИНТЕТИЧЕСКИХ МАТЕРИАЛОВ

ПРОВЕДЕНИЕ САНИТАРНО-ХИМИЧЕСКОГО ИССЛЕДОВАНИЯ ИЗДЕЛИЙ, ИЗГОТОВЛЕННЫХ ИЗ ПОЛИМЕРНЫХ И ДРУГИХ СИНТЕТИЧЕСКИХ МАТЕРИАЛОВ ИЛИ С ИХ ПРИМЕНЕНИЕМ
1. Характеристика исследуемого образца
а) цвет наружной и внутренней поверхностей;
б) поверхность образца (гладкая, шероховатая, неровная и т.д.);
в) запах образца.
Результаты определения выражают описательно с указанием: а) характера запаха (фенольный, ароматический, посторонний, неприятный и т.д.) и б) интенсивность запаха выражают в баллах, пользуясь таблицей.
В случае наличия запаха интенсивностью выше 1 балла образец без дальнейших исследований считают непригодным для применения в пищевой промышленности и быту. При наличии запаха интенсивностью до 1 балла образец подвергается дальнейшему исследованию.
2. Подготовка образца к исследованию
Образец посуды, тары и т.д. после внешнего осмотра моют с помощью кусочка марли теплой водопроводной, а затем дистиллированной водой.
Упаковочные материалы, предназначенные для затаривания пищевых продуктов с влажностью выше 15%, исследуют в виде квадратов 4 x 5 см; загрязнения, могущие быть на поверхности в виде пыли, удаляют путем погружения каждого квадрата последовательно в два стакана с дистиллированной водой и тут же помещают в соответствующие модельные растворы (пищевые кислоты, поваренную соль, дистиллированную воду и др.).
Исследование целлофана проводится без предварительной мойки.
Исследование изделий, предназначенных для контакта с сухими пищевыми продуктами (с влажностью до 15%)
При этих исследованиях используется способность пищевых продуктов сорбировать летучие вещества; кроме того, проводится определение летучих веществ, выделяемых образцом в воздушную среду. В качестве сорбента применяют хлеб, печенье, муку, масло и другие пищевые продукты, исходя из условий эксплуатации образца на практике. Исследуемый образец емкости после вышеуказанной подготовки вытирают чистым сухим полотенцем и помещают в него тот или иной продукт, закрывают крышкой или стеклянной пластинкой; при исследовании отдельных деталей образец вместе с пищевым продуктом помещают в эксикатор или другую герметически закрывающуюся стеклянную емкость. При этом поверхность образца должна быть 3000 кв. см и объем эксикатора 7,5 литра. Условия опыта устанавливают исходя из наиболее неблагоприятных условий, встречающихся при эксплуатации исследуемых изделий на практике. Время экспозиции образца такое же, как и для изделий, предназначенных для контакта с пищевыми продуктами с влажностью выше 15%. Одновременно пищевые продукты помещают в стеклянную банку или эксикатор без образца (контроль), закрывают крышкой и выдерживают в аналогичных условиях. После соответствующей экспозиции проводят закрытую дегустацию пищевого продукта, контактировавшего с образцом, пользуясь для сравнения пищевым продуктом, являющимся контролем.
Лимитирующим показателем при гигиенической оценке образца служат данные, полученные при органолептических исследованиях пищевых продуктов.
В случае изменения органолептических свойств пищевых продуктов (цвет, запах и вкус) исследуемый образец признается непригодным для использования по назначению. Если органолептика пищевого продукта (сорбента) остается без изменения, образец подвергают дальнейшему исследованию.
Исследуемый образец с общей площадью 3000 кв. см помещают в стеклянную емкость объемом 7,5 литра (соотношение площади образца к объему воздуха 1:2,5).
Стеклянная емкость должна иметь две отводные трубки: одну — доходящую до дна, вторую — оканчивающуюся под пробкой с таким расчетом, чтобы при взятии пробы протягиваемый воздух проходил через всю емкость. Экспозиция и температурный режим при данном испытании остаются такими же, как и при испытании образцов, предназначенных для контакта с пищевыми продуктами с влажностью свыше 15%. После соответствующей экспозиции через емкость с образцом протягивают предварительно очищенный воздух <*> со скоростью, указанной в методе определения искомого вещества, и улавливают летучие вещества в два последовательно соединенных поглотительных прибора, содержащих соответствующий поглотительный раствор.
<*> Для очистки протягиваемого воздуха используют нелетучие растворы химических веществ, способные задерживать содержащиеся в воздухе лаборатории вещества, мешающие проведению исследования. Перед поступлением в емкость с исследуемым образцом очищенный воздух должен быть освобожден от паров воды путем протягивания его через прокаленный хлористый кальций или концентрированную серную кислоту.
В некоторых случаях, например, могут быть использованы: 10% раствор щелочи (1-й поглотитель), 10% раствор марганцовокислого калия, подкисленного серной кислотой (2-й поглотитель), концентрированная серная кислота (3-й поглотитель), после чего воздух поступает в емкость с исследуемым образцом.
При выборе поглотительного раствора исходят из физико-химических свойств определяемого ингредиента, его растворимости в тех или иных растворителях и неспособности к образованию с поглотительными растворами летучих соединений. При этом учитывается также возможность дальнейшего определения искомых ингредиентов в поглотительном растворе.
Количество протягиваемого воздуха должно быть в 10 раз больше того количества, которое находится в емкости с образцом.
В поглотительном растворе определяют отдельные ингредиенты, входящие в рецептуру исследуемого изделия, пользуясь методами, изложенными в настоящей Инструкции; в случае их отсутствия пользуются соответствующими руководствами по санитарно-химическому исследованию воздуха <*>. Количества обнаруженного вещества выражают в мг на 1 куб. м воздуха (X).
<*> М.В. Алексеева. Определение атмосферных загрязнений. М.: «Медгиз», 1963; М.С. Быховская, С.Л. Гинзбург, О.Д. Хализова. Методы определения вредных веществ в воздухе. М.: «Медицина», 1966; Е.А. Перегуд. Санитарная химия полимеров. Л.: «Химия», 1967.
Формула расчета:
где а — количество вещества, найденное в анализируемом объеме раствора из первого поглотительного прибора, в гамма; б — объем раствора, взятый для анализа, в мл; в — объем раствора из первого поглотителя, в мл; V — объем воздуха в эксикаторе, в л.
При обнаружении искомого вещества в растворе из второго поглотительного прибора расчет проводят по этой же формуле и результаты суммируют.
Найденные количества оценивают исходя из допустимых количеств данных веществ в атмосферном воздухе населенных мест.
Исследование изделий, предназначенных для контакта с пищевыми продуктами, имеющими влажность свыше 15%
Исследуемый образец изделия после соответствующей мойки подвергают обработке определенными модельными растворами, выбираемыми в зависимости от того, для контакта с какими пищевыми продуктами предназначается использовать данное изделие. Обработка модельными растворами проводится при определенной экспозиции, температурном режиме и с учетом площади поверхности образца. Если исследуемый образец невелик по объему, его помещают в плотно закрывающуюся стеклянную емкость и заливают модельными растворами таким образом, чтобы образец был полностью погружен в них. Если образец велик, то модельные растворы наливают в него и плотно закрывают. Особое внимание следует обратить на герметизацию в тех случаях, когда можно предполагать выделение из синтетического материала летучих компонентов.
В том случае, когда образец полностью погружен в модельные растворы, рассчитывают площадь его внутренней и наружной поверхности. Если же модельные растворы контактируют лишь с внутренней поверхностью, то ведут расчет площади, покрытой жидкостью.
Площадь поверхности образца рассчитывают по обычным геометрическим формулам с известным приближением.
Упаковочные пленки, пластинки, покрытые лаком, шпаклевкой и т.п., помещают в плотно закрывающийся стеклянный сосуд и заливают модельными растворами из расчета на 2 кв. см поверхности 1 куб. см модельного раствора (с учетом площади обеих поверхностей).
Дублированные пленки исследуются в виде мешочков, в которые наливаются модельные растворы.
Полученные результаты анализа пересчитывают в мг/л с указанием площади, контактировавшей с модельным раствором (в кв. см), и количества модельного раствора, взятого для обработки изделия (в мл).
Пример. Изделие из мелалита с площадью, равной 300 кв. см, обработано 1% раствором уксусной кислоты в количестве 150 мл (V).
Содержание формальдегида в мл/г вытяжки (X) определяют по формуле:
где C — количество формальдегида, найденное в анализируемом объеме раствора, в гамма; V — количество исследуемого раствора (вытяжки), взятое для определения, в мл.
Площадь образца, контактировавшего с уксусной кислотой, равна 300 кв. см; количество модельного раствора, взятого для обработки вышеуказанной поверхности, — 150 мл.
Полученные данные (в мг/л) сопоставляют с допустимыми количествами веществ, мигрирующих из изделий в модельные растворы.
На основании всего комплекса данных, полученных при исследовании образца (органолептических, полученных при исследовании образца и вытяжек из него, миграции в вытяжки общего количества органических веществ, отдельных ингредиентов и т.д.), делают заключения о пригодности его для контакта с пищевыми продуктами.
ПЕРЕЧЕНЬ МОДЕЛЬНЫХ РАСТВОРОВ, ИСПОЛЬЗУЕМЫХ ПРИ ИССЛЕДОВАНИИ ИЗДЕЛИЙ ИЗ СИНТЕТИЧЕСКИХ МАТЕРИАЛОВ
| Наименование продуктов, для контакта с которыми предназначены изделия | Модельные растворы, имитирующие пищевые продукты |
| Мясо, рыба свежие | Дистиллированная вода, 0,3% раствор молочной кислоты |
| Мясо и рыба соленые и копченые | Дистиллированная вода, 5% раствор поваренной соли |
| Молоко, молочнокислые продукты и молочные консервы | Дистиллированная вода, 0,3% раствор молочной кислоты, 3,0% раствор молочной кислоты |
| Колбаса вареная; консервы: мясные, рыбные, овощные; овощи маринованные и квашеные, томат-паста и др. | Дистиллированная вода, 2% раствор уксусной кислоты, содержащей 2% поваренной соли; нерафинированное подсолнечное масло |
| Фрукты, ягоды, фруктово-овощные соки, консервы фруктово-ягодные, безалкогольные напитки, пиво | Дистиллированная вода, 2% раствор лимонной кислоты |
| Алкогольные напитки, вина | Дистиллированная вода, 20% раствор этилового спирта, 2% раствор лимонной кислоты |
| Водки, коньяки | Дистиллированная вода, 40% раствор этилового спирта |
| Спирт пищевой, ликеры, ром | Дистиллированная вода, 96% раствор этилового спирта |
| Готовые блюда и горячие напитки (чай, кофе, молоко и др.) | Дистиллированная вода, 1% раствор уксусной кислоты |
Примечания.
1. Изделия, используемые в условиях, отличных от вышеизложенных, обрабатываются при максимальном приближении к режимам эксплуатации с некоторой аггравацией.
2. При исследовании изделий из пластмасс, содержащих азот и альдегиды, в качестве модельной среды используют лимонную кислоту 0,3% и 3% вместо молочной кислоты.
3. Пункт утратил силу. (в ред. Методических указаний, утв. заместителем Главного государственного санитарного врача СССР 30.06.87 N 4395-87)
Моделирование продолжительности контакта изделий с модельными растворами
Продолжительность контакта изделия с модельными растворами устанавливается в зависимости от условий эксплуатации его с некоторой аггравацией:
а) если время предполагаемого контакта пищевого продукта с изделием не превышает 10 минут, экспозиция при исследовании — 2 часа;
б) если время контакта пищевого продукта с изделием не превышает 2 часов, экспозиция при исследовании — 1 сутки;
в) если время контакта пищевого продукта с изделием от 2 до 48 часов, экспозиция при исследовании — 3 суток;
г) если время контакта пищевого продукта с изделием свыше 2 суток, экспозиция при исследовании — 10 суток;
д) подпункт утратил силу. (в ред. Методических указаний, утв. заместителем Главного государственного санитарного врача СССР 30.06.87 N 4395-87)
е) изделия, предназначенные для контакта с пищевыми продуктами, подлежащими стерилизации, наполняют модельными растворами и автоклавируют в герметически закрытом виде в течение 2 часов и далее оставляют на 10 суток при комнатной температуре.
Температурные режимы при исследовании изделий
а) Изделия, предназначенные для контакта с пищевыми продуктами при температуре окружающей среды, заливают модельными растворами комнатной температуры и выдерживают в течение вышеуказанного времени;
б) изделия, предназначенные для контакта с горячей пищей (столовая, чайная, кофейная посуда), заливают нагретыми до 80 град. модельными растворами и далее выдерживают при комнатной температуре в течение вышеуказанного времени;
в) изделия и упаковочные материалы, предназначенные для затаривания пищевых продуктов в горячем виде (топленое масло, плавленые сыры и др.), заливают модельными растворами с температурой 80 град. и далее выдерживают при комнатной температуре в течение вышеуказанного времени;
г) автоклавирование проводят при 121 град.;
д) формы для выпечки хлеба, ветчины и т.д. заливают кипящим модельным раствором, закрывают крышкой и кипятят в течение часа.
Органолептическое исследование вытяжек, полученных после соответствующей обработки изделий
Органолептические свойства вытяжек из исследуемых изделий обусловливаются переходом в них веществ, входящих в рецептуру исследуемого изделия. Органолептические свойства вытяжек являются одним из важных показателей при санитарно-химическом исследовании изделий из полимерных материалов, поэтому определение их должно проводиться со всей ответственностью. Во избежание ошибок органолептическое испытание проводят комиссионно (несколько человек — не менее пяти) методом закрытой дегустации.
В дегустации могут участвовать только те лица, которые четко различают запах, вкус и привкус в образцах. В связи с этим необходимо провести отбор дегустаторов следующим способом: для дегустации предлагают два одинаковых контрольных образца и два одинаковых образца, имеющих слабый посторонний запах, вкус или привкус. Лица, обнаружившие несколько раз различие в органолептике между одинаковыми образцами, не могут участвовать в дегустации.
При органолептическом исследовании вытяжек определяют наличие мути, осадка, постороннего запаха, вкуса или привкуса.
1. Мутность вытяжек характеризуют описательно: слабая опалесценция, опалесценция, сильная опалесценция, слабая муть, заметная муть, сильная муть.
2. Осадок характеризуют по его величине: ничтожный, незначительный, заметный, большой. Кроме того, отмечают его свойства: кристаллический, аморфный и т.п.; отмечают также цвет осадка: белый, серый, бурый и т.п.
3. Запах и его интенсивность определяют сразу же после окончания соответствующей экспозиции во всех вытяжках из исследуемого образца при комнатной температуре, а в водной вытяжке и после нагревания приблизительно до 60 град. C.
При определении запаха при комнатной температуре исследуемые вытяжки и контрольные модельные растворы должны иметь комнатную температуру.
Если температурные условия обработки образца отличаются от комнатной, проводят определение запаха также и при комнатной температуре.
Определение запаха в вытяжках
Определение запаха в вытяжках проводят путем закрытой дегустации, исключающей обмен мнениями между дегустаторами, методом «расширенного треугольника».
Определение запаха при комнатной температуре. В четыре колбы Эрленмейера с притертыми пробками емкостью по 100 мл вносят: в три колбы — по 50 мл контрольного модельного раствора и в одну — 50 мл исследуемого раствора и закрывают пробками.
Предварительно каждому дегустатору предлагают открыто ознакомиться с запахом контрольного модельного раствора. Для этого одну из трех колбочек с контрольным модельным раствором тщательно взбалтывают, открывают пробку и слегка втягивают в нос воздух из колбочки у самого горлышка.
После этого проводят закрытую дегустацию растворов в оставшихся трех колбочках, чтобы выявить запах раствора, отличающийся от контрольного.
Характер запаха выражают описательно, например: фенольный, ароматический, посторонний неопределенный и т.д.
Интенсивность запаха выражают в баллах.
Каждый дегустатор результаты исследования заносит в индивидуальную дегустационную карту.
Из полученных от каждого дегустатора результатов определения интенсивности запаха выводят ее среднее арифметическое значение.
Пример. Дегустаторы определили наличие запаха интенсивностью 0, 1, 2,2 и 1 балл. Среднее арифметическое равно 1,2 балла. Десятые доли до 0,5 отбрасываются, а от 0,5 и более — округляют до целого значения следующего балла. В нашем случае интенсивность запаха будет равна 1 баллу.
Определение запаха в водной вытяжке при нагревании. В четыре колбы Эрленмейера объемом по 100 мл вносят: в три колбы — по 50 мл дистиллированной воды, которая использовалась для получения вытяжек из образца (контроль), в четвертую — 50 мл исследуемой водной вытяжки. Колбы закрывают хорошо подобранными часовыми стеклами и нагревают на водяной бане приблизительно до 60 град. C, взбалтывают содержимое колб вращательными движениями, сдвигают часовое стекло в одну сторону и быстро определяют запах.
Вначале каждому дегустатору предлагают открыто ознакомиться с запахом нагретого контрольного модельного раствора, как указано выше. Затем проводят закрытую дегустацию нагретых модельных растворов в оставшихся трех колбочках, чтобы выявить запах раствора, отличающийся от контрольного.
Характер и интенсивность запаха обозначают так же, как и при определении запаха в вытяжках при комнатной температуре.
4. Вкус и привкус определяют только в водных вытяжках из исследуемого изделия при комнатной температуре и при температуре около 40 град. C по сравнению с контролем методом закрытой дегустации аналогично определению запаха. При этом набирают в рот 10 — 15 мл контрольной воды, держат во рту несколько секунд не проглатывая, а затем сплевывают, точно так же поступают с исследуемыми растворами.
Привкус характеризуют словами: горьковатый, щиплющий, нефтепродуктов, посторонний неопределенный и т.д. Интенсивность привкуса выражают словами: слабый привкус, ясно выраженный, сильный.
Оценка образцов на основании органолептических исследований
При наличии одного из вышеперечисленных изменений органолептических свойств вытяжек: запаха выше 1 балла, постороннего привкуса (обнаруживаемого всеми дегустаторами), наличии мути, осадка, изменения цвета вытяжки — образец признается непригодным для использования в пищевой промышленности.
В случае отсутствия органолептических изменений проводят химическое исследование вытяжек исходя из рецептуры образца.
Таблица 1
ОПРЕДЕЛЕНИЕ ИНТЕНСИВНОСТИ ЗАПАХА
| Интенсивность запаха (балл) | Характеристика | Проявление запаха |
| 0 | Никакого запаха | Отсутствие ощутимого запаха |
| 1 | Очень слабый | Запах, обычно не замечаемый, но обнаруживаемый опытным исследователем |
| 2 | Слабый | Запах, обнаруживаемый неопытным дегустатором, если обратить на это его внимание |
| 3 | Заметный | Запах, легко замечаемый и могущий вызвать неодобрительный отзыв |
| 4 | Отчетливый | Запах, обращающий на себя внимание, вызывающий отрицательный отзыв |
| 5 | Очень сильный | Запах настолько сильный, что вызывает неприятное ощущение |
ПРИМЕРНЫЙ ПЕРЕЧЕНЬ КОМПОНЕНТОВ, КОТОРЫЕ СЛЕДУЕТ ОПРЕДЕЛЯТЬ ПРИ САНИТАРНО-ХИМИЧЕСКОМ ИССЛЕДОВАНИИ ИЗДЕЛИЙ ИЗ ПОЛИМЕРНЫХ МАТЕРИАЛОВ ИСХОДЯ ИЗ РЕЦЕПТУРЫ
| Наименование полимерных материалов | Рецептура | Определение веществ, имеющих гигиеническое значение |
| 1 | 2 | 3 |
| Анид (полиамид на основе гексаметилендиамина и адипиновой кислоты) | Гексаметилендиамин Адипиновая кислота Соль АГ (адипиновокислый гексаметилендиамин) |
Общее количество органических веществ Гексаметилендиамин |
| Винипласт (непластифицированный поливинилхлорид) | Поливинилхлорид Стеарат кальция Стеарат цинка Двуокись титана Эпоксидированное растительное масло |
Общее количество органических веществ Бромирующиеся вещества Цинк Хлориды |
| Декоррозит (фенолформальдегидный пресс-порошок) | Фенолформальдегидная смола, поливинилхлоридная смола, стабилизатор — гексаметилентетрамин (уротропин), стеарат кальция, известь, кокс | Общее количество органических веществ Фенол Формальдегид Хлориды Гексаметилентетрамин |
| Капрон | Е-капролактам Уксусная кислота Титана двуокись |
Общее количество органических веществ Е-капролактам |
| Лак 71 | Фенол Ксиленол Формальдегид Фталевый ангидрид Глифталовая смола N 135 |
Общее количество органических веществ Фенолы (фенол и ксиленол) Формальдегид Фталевый ангидрид |
| Меладур (пресс-материал) | Меламин Формальдегид Титана двуокись Клетчатка бука |
Общее количество органических веществ Меламин Формальдегид |
| Мелалит (пресс-порошок) | Меламин Формальдегид Целлюлоза сульфитная Двуокись титана Пигмент голубой фталоцианиновый |
То же |
| Нейлон (полиамид на основе гексаметилендиамина и адипиновой кислоты) | Гексаметилендиамин Адипиновая кислота Соль АГ (адипиновокислый гексаметилендиамин) |
Общее количество органических веществ Гексаметилендиамин |
| Органическое стекло (плексиглас полиметилметакрилат) | Метилметакрилат Дибутилфталат Перекись бензоила |
Общее количество органических веществ Бромирующиеся вещества Метилметакрилат Дибутилфталат Метиловый спирт (образующийся в процессе деструкции) |
| Перлон (полиамид на основе Е-капролактама) | Е-капролактам Уксусная кислота Титана двуокись |
Общее количество органических веществ Е-капролактам |
| Пленка перфоль ПК-4 (полиамид на основе Е-капролактама) | То же | То же |
| Полиамидные смолы на основе Е-капролактама (поликапролактам, иначе называемый капрон или перлон, пленка перфоль ПК-4) | Е-капролактам Уксусная кислота Титана двуокись |
Общее количество органических веществ Е-капролактам |
| Полиамидные смолы на основе гексаметилендиамина и адипиновой кислоты (анид, нейлон) | Гексаметилендиамин Адипиновая кислота Соль АК (адипиновокислый гексаметилендиамин) |
Общее количество органических веществ Гексаметилендиамин |
| Поливинилхлорид непластифицированный (см. винипласт) | ||
| Поливинилхлорид пластифицированный | Полихлорвиниловая смола Дибутилфталат Стеарат кальция |
Общее количество органических веществ Бромирующиеся вещества Дибутилфталат Хлориды |
| Полиизобутилен | Общее количество органических веществ Бромирующиеся вещества |
|
| Полиметилметакрилат (см. органическое стекло) | ||
| Полипропилен | Пропилен Триэтилалюминий Титан четыреххлористый Бензин Изопропиловый спирт |
Общее количество органических веществ Бромирующиеся вещества Формальдегид (образующийся в процессе деструкции) |
| Политетрафторэтилен (фторопласт) | Тетрафторэтилен Персульфат калия Бура |
Общее количество органических веществ Бромирующиеся вещества Фтор |
| Полистирол 1. Блочный «Т» | Стирол | Общее количество органических веществ Бромирующиеся вещества Стирол |
| 2. Суспензионный «ПС-С» | Стирол Стеарат цинка Тетрабутилпербензоат Перекись бензоила |
Общее количество органических веществ Бромирующиеся вещества Стирол Цинк |
| Полистирол других марок | Общее количество органических веществ Бромирующиеся вещества Стирол Другие ингредиенты исходя из рецептуры |
|
| Полиэтилен низкой плотности (высокого давления) нестабилизированный | Получается полимеризацией этилена при высоком давлении | Общее количество органических веществ Бромирующиеся вещества |
| Полиэтилен высокой плотности (низкого давления) | Этилен Триэтилалюминий Титан четыреххлористый Бензин Изопропиловый спирт |
Общее количество органических веществ Бромирующиеся вещества |
| Резит (отвержденная резольно-фенолформальдегидная смола) | Фенол Формальдегид Едкий натр |
Общее количество органических веществ Фенол Формальдегид |
| Смола бесфенольная фенолформальдегидная | Фенол Формальдегид Едкий натр |
Общее количество органических веществ Фенол Формальдегид |
| Смола марки «СП-3» | Фенол Формальдегид Едкий натр Аммиак Желатина (или декстрин) Натрий хлористый Уксусная кислота Спирт |
Общее количество органических веществ Фенол Формальдегид |
| Энант (продукт конденсации аминоэнантовой кислоты) | Общее количество органических веществ Азот Хлориды |
|
| Эпоксидное покрытие металлических емкостей на основе смол ЭД-5 и ЭД-6 | ||
| 1. Первый слой (грунт ЭС) | Эпоксидная смола ЭД-5: эпихлоргидрин, дифенилолпропан, едкий натр Железный сурик Полиэтиленполиамин Ацетон |
Общее количество органических веществ Бромирующиеся вещества Эпихлоргидрин Дифенилолпропан Полиэтиленполиамин (см. определение общего количества аминосоединений) |
| 2. Второй слой (грунт ЭК) | Эпоксидная смола ЭД-5: эпихлоргидрин, дифенилолпропан, едкий натр Каолин Полиэтиленполиамин |
|
| 3. Эмаль ЭТК (наносится на грунт ЭК) | Эпоксидная смола ЭД-6: эпихлоргидрин, дифенилолпропан, едкий натр Двуокись титана Ацетон Полиэтиленполиамин |
Примечание. Окисляемость и бромирующиеся вещества являются общеориентировочными показателями, которые не нормируются.
ХИМИЧЕСКОЕ ИССЛЕДОВАНИЕ ВЫТЯЖЕК, ПОЛУЧЕННЫХ ПОСЛЕ СООТВЕТСТВУЮЩЕЙ ОБРАБОТКИ ИЗДЕЛИЙ
ОПРЕДЕЛЕНИЕ ОБЩЕГО КОЛИЧЕСТВА ОРГАНИЧЕСКИХ ВЕЩЕСТВ В ВОДНОЙ ВЫТЯЖКЕ ПО ИХ ОКИСЛЯЕМОСТИ
Окисляемостью называется количество кислорода, выраженное в миллиграммах, которое необходимо для окисления неорганических и органических веществ.
Если в рецептуру изделия входят неорганические восстановители, их необходимо определить специальными методами и расход окислителя, соответствующий содержанию неорганических веществ, вычесть из общей окисляемости. По разности можно приблизительно установить содержание органических веществ, перешедших из исследуемого изделия в водную вытяжку.
Таким образом, определением окисляемости можно приблизительно установить содержание органических веществ, для чего, главным образом, это определение и проводится.
В зависимости от применяемого окислителя различают следующие методы определения окисляемости: перманганатный, бихроматный, цериевый, йодатный и хлорный (хлорное число).
Результаты, полученные различными методами, могут быть различны для одной и той же пробы вследствие неодинаковой степени окисления органических веществ, которая зависит от свойств окислителя и его концентрации, температуры, pH и т.д.
В настоящее время еще нет методов, с помощью которых можно было бы полностью окислить все встречающиеся органические вещества.
Перманганатный метод в двух его вариантах — окисление в кислой среде или в щелочной среде — очень неточен не только потому, что окисление органических веществ перманганатом проходит неполно и многие из них совсем не окисляются, но и потому, что и сам окислитель — перманганат калия — в принятых условиях метода в некоторой степени разлагается.
Йодатный метод не имеет существенных преимуществ по сравнению с бихроматным, но он требует значительно больше времени и, как показала практика, иногда дает плохо воспроизводимые результаты.
В связи с этим ниже приводится описание только бихроматного метода определения окисляемости, предложенного для определения органических веществ при исследовании воды.
Определение окисляемости бихроматным методом
Бихромат при кипячении в сернокислой среде окисляет большинство органических веществ. Избыток бихромата определяется титрованием. Для повышения полноты окисления органических веществ добавляют сульфат серебра в качестве катализатора.
Аппаратура
Колбы круглодонные со шлифами на 300 мл.
Обратные холодильники, пришлифованные к этим колбам.
Реактивы
1. Бихромат калия, 0,25-н основной раствор 12,258 г K2Cr2O7 ч.д.а., предварительно высушенного в течение 2 часов при 105 град. C, растворяют в дистиллированной воде и доводят до 1 л.
2. Бихромат калия, 0,025-н раствор: 100 мл 0,25-н раствора бихромата калия разбавляют дистиллированной водой до 1 л.
3. Серная кислота ч.д.а., концентрированная с уд. в. 1,84 (96%). Для окисления возможной примеси органических веществ концентрированную серную кислоту наливают в колбу Кьельдаля, закрывают воронкой и кипятят в течение 4 часов; после охлаждения сливают в хорошо вымытую хромовой смесью и высушенную стеклянную банку с притертой пробкой, закрывают сверху колпачком из плотной бумаги и для защиты от внешнего загрязнения хранят под стеклянным колпаком.
4. Сернокислое серебро ч.д.а., кристаллическое. Приготовление: 34 г азотнокислого серебра (ч.д.а.) растворяют в 20 мл горячей бидистиллированной воды, добавляют предварительно профильтрованный через стеклянную воронку с пористым дном горячий раствор 13,2 г сернокислого аммония (NH4)2SO4 (ч.д.а.) в 20 мл воды. По охлаждении кристаллический осадок Ag2SO4 отсасывают в стеклянной воронке с пористым дном, промывают холодной водой и сушат в сушильном шкафу.
5. Соль Мора (двойная соль сернокислого закисного железа и сернокислого аммония) — Fe(NH4)2(SO4)2 x 6H2O (ч.д.а.), 0,25-н раствор: 98 г соли Мора растворяют в дистиллированной воде, добавляют 20 мл концентрированной серной кислоты и после охлаждения доводят дистиллированной водой до 1 л. Титр раствора устанавливают для каждой серии определений, для чего 25 мл 0,25-н раствора бихромата калия разбавляют дистиллированной водой приблизительно до 250 мл, прибавляют 20 мл концентрированной серной кислоты и после охлаждения титруют раствором соли Мора при добавлении 3 — 4 капель раствора ферроина или дифениламина (или 10 — 15 капель раствора N-фенилантраниловой кислоты или дифениламинсульфоната натрия).
6. Соль Мора, 0,025-н титрованный раствор. Приготовляют соответствующим разбавлением 0,25-н раствора.
Титр устанавливают так же, как и 0,25-н раствора, используя 0,025-н раствор бихромата калия.
7. Индикатор, один из следующих растворов: а) ферроин — 1,485 г моногидрата, 1,10-фенантролина и 0,695 г FeSO4 x 7H2O (ч.д.а.) растворяют в дистиллированной воде и доводят до 100 мл; б) дифениламин — 1 г дифениламина растворяют в 100 мл концентрированной серной кислоты; в) N-фенилантраниловая кислота — 0,25 г растворяют в 12 мл 0,1-н раствора едкого натра и разбавляют водой до 250 мл; г) дифениламинсульфонат натрия или бария — 0,2% водный раствор.
8. Бидистиллированная вода. К дистиллированной воде добавляют марганцовокислый калий до малинового окрашивания и 1 мл концентрированной серной кислоты, после чего проводят вторичную перегонку в дистилляционном аппарате со стеклянными шлифами.
Ход определения
50 мл водной вытяжки помещают в круглодонную колбу на 300 мл с обратным холодильником. Добавляют 25 мл 0,025-н раствора бихромата калия, 0,4 г сульфата серебра и стеклянные шарики или стеклянные капилляры, запаянные с одного конца, размером около 1 см, перемешивают, осторожно приливают малыми порциями 75 мл концентрированной серной кислоты, смесь тщательно перемешивают после добавления каждой порции, соединяют колбу с обратным холодильником и содержимое колбы нагревают до слабого кипения, которое поддерживают в течение 2 часов. После охлаждения обмывают стенки холодильника 25 мл бидистиллированной воды и переносят содержимое круглодонной колбы в коническую колбу емкостью 500 мл, обмывая стенки круглодонной колбы несколько раз 175 мл бидистиллированной воды. Содержимое колбы охлаждают, добавляют 3 — 4 капли индикатора ферроина или дифениламина, или 10 — 15 капель раствора N-фенилантраниловой кислоты, или 10 — 15 капель дифениламинсульфоната натрия и оттитровывают избыток бихромата титрованным раствором соли Мора до перехода окраски из синевато-зеленой в красновато-синюю, пользуясь при этом микробюреткой с делениями, равными 0,02 мл.
Проводят холостой опыт с 50 мл бидистиллята вместо исследуемой вытяжки в одинаковых условиях с исследуемой вытяжкой.
Расчет
Окисляемость бихроматная в мг кислорода на:
| л = | (а — в) К 0,2 x 1000 | = | (а — в) К 200 | , |
| V | V |
где а — расход титрованного раствора соли Мора на холостой опыт, в мл; в — расход титрованного раствора соли Мора на титрование исследуемой вытяжки, в мл; К — поправочный коэффициент 0,025-н раствора соли Мора; 0,2 — количество мг кислорода (1 мл 0,025-н раствора соли Мора соответствует 0,25 мг кислорода); V — количество исследуемой вытяжки, взятой для определения, в мл.
Примечание. Необходимо иметь в виду, что незначительные загрязнения кислоты, посуды, холодильника и пр. следами органических веществ приводят к грубым ошибкам.
Окисляемость характеризует водоустойчивость исследуемого материала. При наличии органических веществ в водной вытяжке, обнаруженных по их окисляемости, необходимо исследовать вытяжки из исследуемого изделия на переход в них отдельных ингредиентов, входящих в рецептуру полимерного материала.
Нормировать количества органических веществ, обнаруженных в водной вытяжке по их окисляемости, не представляется возможным вследствие различной токсичности отдельных органических веществ. Так, например, высокая окисляемость, полученная при исследовании пленочных материалов, пластифицированных глицерином, не может служить лимитирующим показателем при оценке исследуемого материала, если при этом не изменены органолептические свойства вытяжки. С другой стороны, небольшая окисляемость, обусловленная наличием в водной вытяжке токсичного вещества, например стирола, является отрицательным показателем.
ОПРЕДЕЛЕНИЕ БРОМИРУЮЩИХСЯ ВЕЩЕСТВ
С помощью определения бромирующихся веществ можно получить представления о миграции из изделия из полимерного материала в модельную среду, контактирующую с ним, фенола, непредельных соединений и других веществ, присоединяющих бром, т.е. о суммарном количестве органических веществ, реагирующих с бромом.
Нормировать суммарное количество бромирующих веществ без их разделения не представляется возможным вследствие различной токсичности отдельных бромирующих веществ.
Необходимый для реакции бром получается при взаимодействии бромата калия (KBrO3) с бромидом калия (KBr) в кислой среде:
КВrО3 + 5КВr + 3H2SO4 -> 3Вr2 + 3К2SО4 + 3Н2O.
Обычно или добавляют бромид в анализируемый раствор перед титрованием, или вводят его в титрованный раствор бромата.
Реактивы
1. Бромидброматная смесь. Чтобы получить 0,1-н раствор брома, растворяют точно 2,7837 г чистого, высушенного при 180 град. в течение 1 — 2 часов бромата калия:
| ( | KBrO3 | = | 167,02 | = 2,7837 г), |
| 60 | 60 |
| около 10 г бромида калия ( | 5KBr | = | 5119,02 | = 9,92 г) и доводят до 1 л. |
| 60 | 60 |
Бромат калия (KBrO3) должен быть точно отвешен, тогда как бромид калия (KBr) может быть отвешен на технических весах, но навеска его не должна быть меньше 9,92, избыток бромида калия не мешает.
Перед определением готовят 0,005-н раствор бромидброматной смеси. Для этого 50 мл 0,1-н раствора бромидброматной смеси разбавляют бидистиллированной водой до 1 л.
2. Тиосульфат натрия (Na2S2O3), 0,1-н раствор. Перед употреблением из 0,1-н раствора тиосульфата натрия готовят соответствующим разведением 0,005-н раствор.
3. Йодид калия (KJ).
4. Серная кислота, разведенная 1:3 (по объему).
5. Крахмал, 0,5% раствор, свежеприготовленный.
Ход определения
50 мл вытяжки из исследуемого изделия, не содержащей спирта и сахара <*>, переносят в коническую колбу емкостью 250 — 300 мл с хорошо притертой пробкой и добавляют 25 мл бромидброматной смеси. Затем прибавляют 10 мл разбавленной 1:3 серной кислоты, тут же закрывают колбу пробкой, осторожно перемешивают содержимое колбы и ставят в темное место на 30 минут. Далее добавляют 1 г йодида калия, снова быстро закрывают колбу пробкой, осторожно перемешивают и через 5 минут титруют выделившийся йод 0,005-н раствором тиосульфата натрия до слабо-желтого цвета жидкости. После этого добавляют 1 — 2 мл 0,5% раствора крахмала и продолжают титрование до обесцвечивания раствора.
<*> Спирт и сахар мешают определению.
В другой такой же колбе проводят контрольное определение, для чего вместо исследуемой вытяжки берут 50 мл контрольного модельного раствора и добавляют все реактивы в тех же количествах, в каких они были взяты при исследовании вытяжки, и титруют выделившийся йод 0,005-н раствором тиосульфата натрия.
Результаты определения выражают в количестве прореагировавшего брома (X), в мг/л:
| X = | (а — в) K 1000 x 0,3996 | , |
| г |
где а — объем раствора тиосульфата натрия, израсходованный при проведении контрольного опыта, в мл; в — объем раствора тиосульфата натрия, израсходованный в опыте с вытяжкой из исследуемого материала, в мл; К — поправочный коэффициент для приведения концентрации раствора тиосульфата натрия к точно 0,005-н; 0,3996 — количество мг брома, эквивалентное 1 мл 0,005-н раствора тиосульфата натрия; г — объем вытяжки, взятой для определения, в мл.
ОПРЕДЕЛЕНИЕ ОБЩЕГО КОЛИЧЕСТВА АМИНОСОЕДИНЕНИЙ
Метод основан на количественном определении аминосоединений по общему содержанию азота путем их минерализации по Кьельдалю с последующим определением аммонийного азота колориметрическим методом с реактивом Несслера. Определению мешает аммиак.
Чувствительность метода 0,001 мг азота в колориметрируемом объеме.
Реактивы
Все реактивы готовят на дистиллированной воде, не дающей положительной реакции на аммиак.
1. Серная кислота, концентрированная, химически чистая, прокипяченная для сожжения органических веществ и проверенная на отсутствие в ней аммиака.
2. Медь сернокислая, 10% раствор.
3. Калий сернокислый, 10% раствор.
4. Безаммиачная дистиллированная вода.
Безаммиачную воду получают вторичной перегонкой обычной дистиллированной воды, подкисленной 1 мл концентрированной серной кислоты с добавлением перманганата калия до малинового окрашивания.
5. Кали едкое, 10% раствор.
6. Едкий натр (или калий), концентрированный раствор (500 г едкого натра в 1 л воды).
7. Серная кислота, 0,1-н раствор.
8. Реактив Несслера — 50 г йодата калия растворяют в 50 мл безаммиачной воды; 30 г хлорной ртути (сулема) растворяют при постоянном помешивании в 150 мл безаммиачной воды, нагретой до кипения. Колба с нагретой до кипения водой должна быть перед внесением сулемы снята с нагревательного прибора. Горячий раствор сулемы приливают по каплям к раствору йодата калия до появления нерастворимого красного осадка и оставляют стоять до следующего дня.
Растворяют при нагревании 150 г химически чистого кали едкого (или 107 г едкого натра) в 300 мл безаммиачной воды и по охлаждении вливают его в раствор йодистого калия и хлорной ртути, доводят объем до 1 л безаммиачной водой и добавляют сверх этого объема 4 — 6 мл раствора сулемы до появления неисчезающего красного осадка.
Реактив оставляют стоять в темном месте до полного осветления и в дальнейшем хранят также в темноте хорошо закрытым (но не стеклянной пробкой). При употреблении берут прозрачный раствор, не взмучивая осадка. Хорошо отстоявшийся раствор слить сифоном с осадка и в таком виде хранить.
Все операции по приготовлению реактива Несслера следует выполнять в вытяжном шкафу.
9. Стандартный раствор хлористого аммония.
0,3818 г х.ч. хлористого аммония растворяют в безаммиачной воде и доводят объем раствора до 100 мл (1 мл = 1 мг азота). Раствор должен быть всегда свежеприготовленным. Разведением в 100 раз основного раствора готовят рабочий стандартный раствор (1 мл = 0,01 мг азота).
Ход определения
50 мл испытуемой вытяжки <*> помещают в колбу Кьельдаля емкостью 250 мл, добавляют 10 мл концентрированной серной кислоты (уд. в. 1,835 — 1,84), 2 — 3 капли 10% раствора сернокислой меди и 1 — 2 мл 10% сульфата калия, кипятят до тех пор, пока раствор в колбе станет вполне прозрачным и бесцветным или слабо-зеленоватым. После окончания сжигания и охлаждения раствора содержимое колбы Кьельдаля без потерь переносят в колбу перегонного аппарата емкостью около 500 мл, применяя при этом приблизительно 200 мл безаммиачной дистиллированной воды.
<*> Вытяжки должны быть получены на безаммиачной воде.
Собирают прибор для отгонки <*>; нижний конец холодильника погружают в мерную колбу (приемник) объемом 200 мл, содержащую 10 мл 0,1-н серной кислоты. Затем, сняв колбу для отгонки аммиака с перегонного прибора, бросают в нее кусочек лакмусовой бумажки и, держа колбу наклонно, осторожно, чтобы жидкости не смешивались, вливают 50 мл концентрированного раствора едкого натра. Не встряхивая содержимого колбы, вводят стеклянные бусинки, сейчас же соединяют колбу с собранной установкой для отгонки, осторожно вращая колбу, смешивают в ней оба слоя жидкости (жидкость должна быть щелочной по лакмусу) и начинают нагревание.
<*> Отгонка аммиака проводится в приборе с пришлифованными стеклянными частями. Прибор предварительно освобождается от возможных следов аммиака. Для этого в него вливают дистиллированную воду и кипятят до исчезновения в отгоне следов аммиака. Оставшуюся в колбе прибора воду выливают.
Отгоняют в приемник с кислотой около 200 мл дистиллята. Содержимое приемной колбы доводят до метки, перемешивают и определяют в нем аммиак с реактивом Несслера.
Для этого в колориметрическую пробирку вносят определенное количество дистиллята в зависимости от содержания азота, прибавляют 1 каплю 10% раствора едкого натра или кали и доводят объем раствора до 10 мл безаммиачной дистиллированной водой.
Затем готовят стандартную шкалу, как указано в табл. 2.
Таблица 2
СТАНДАРТНАЯ ШКАЛА ДЛЯ ОПРЕДЕЛЕНИЯ АЗОТА
| Реактив | Номер пробирки | ||||||
| 0 | 1 | 2 | 3 | 4 | 5 | 6 | |
| Стандартный раствор хлористого аммония (1 мл = 0,01 мг азота), в мл | 0 | 0,1 | 0,2 | 0,4 | 0,6 | 0,8 | 1,0 |
| Дистиллированная вода, в мл | 10,0 | 9,9 | 9,8 | 9,6 | 9,4 | 9,2 | 9,0 |
| Содержание азота, в мг | 0 | 0,001 | 0,002 | 0,004 | 0,006 | 0,008 | 0,01 |
Далее одновременно в пробирку с дистиллятом и стандартным раствором добавляют по 0,2 мл реактива Несслера и тщательно перемешивают.
Через 5 — 10 минут сравнивают интенсивность образовавшейся окраски с окраской стандартных растворов. Расчет производится в мг азота на 1 л вытяжки. Необходимо обязательно ставить контрольный опыт с применяемыми реактивами в тех же условиях.
ОПРЕДЕЛЕНИЕ АКРИЛОНИТРИЛА В ПРИСУТСТВИИ АММИАКА
Метод основан на омылении акрилонитрила до аммиака при нагревании со щелочью и последующем определении аммиака колориметрическим методом с реактивом Несслера.
При взаимодействии аммиака с реактивом Несслера образуется соединение, окрашенное в желтовато-бурый цвет, интенсивность окраски прямо пропорциональна концентрации аммиака.
Реактивы и аппаратура
Дистиллированная вода, не содержащая иона NH+ 4.
Для очистки к 1 л дистиллированной воды прибавляют 5 мл 10% серной кислоты и перегоняют ее. Первые порции отгона (100 мл) отбрасывают. Следующие порции проверяют с реактивом Несслера на содержание иона NH+ 4. При отрицательной реакции на ион NH+ 4 воду собирают и хранят в колбе с нормальным шлифом. Все нижеуказанные растворы готовят на воде, не содержащей аммиака.
Стандартный раствор аммиака <*>.
<*> При определении аммиака непосредственно в водных вытяжках стандартные растворы аммиака готовят на 0,1-н растворе серной кислоты.
1. Основной стандартный раствор, содержащий 0,1 мг/мл аммиака, готовят растворением 0,0314 г хлорида аммония (х.ч.) в 100 мл дистиллированной воды.
2. Рабочий стандартный раствор, содержащий 0,01 мг/мл аммиака, готовят перед определением, разбавляя в 10 раз основной раствор дистиллированной водой.
Стандартный раствор акрилонитрила.
1. Основной раствор: в мерную колбу на 25 мл наливают 10 — 15 мл 0,1-н раствора серной кислоты и колбу взвешивают на аналитических весах. Затем вносят в колбу 2 — 3 капли акрилонитрила и вновь взвешивают. По разности веса определяют количество внесенного акрилонитрила. Раствор в колбе доводят до метки 0,1-н раствором серной кислоты и высчитывают содержание акрилонитрила в 1 мл раствора.
2. Путем соответствующего разведения основного раствора 0,1-н серной кислотой готовят перед употреблением рабочий стандартный раствор с содержанием акрилонитрила 0,0312 мг/мл, что соответствует 0,01 мг аммиака в мл.
Серная кислота — 1,0; 0,7; 0,1-н растворы из фиксанала разбавлением безаммиачной дистиллированной водой.
Едкое кали, 40% раствор.
Реактив Несслера. 10 г йодида ртути тщательно растирают в ступке, добавляют 7 г йодида калия и вторично тщательно растирают смесь порошков, добавляют 3 — 5 мл воды и снова растирают смесь до получения прозрачного раствора зеленоватого цвета. Раствор при непрерывном помешивании переносят малыми порциями в 40% раствор щелочи (16 г KOH в 50 мл безаммиачной дистиллированной воды). Общий объем раствора доводят водой до 100 мл и оставляют на 1 — 2 дня, после чего осторожно сифонируют прозрачный раствор в склянку из темного стекла и закрывают резиновой (корковой) пробкой, обернутой полиэтиленовой пленкой.
Колориметрические пробирки (высотой 15 см) с нормальным шлифом, снабженные воздушными холодильниками (длиной 50 см).
Фотоэлектроколориметр ФЭК-Н-57.
Перегонная установка с нормальными шлифами для концентрирования вытяжки.
Ход определения
Ниже приводятся два метода определения аммиака и акрилонитрила при их совместном присутствии.
1. Аммиак и акрилонитрил определяют непосредственно в водных вытяжках, содержащих значительные количества этих соединений.
Чувствительность метода по аммиаку 0,3 мг/л.
2. Аммиак и акрилонитрил определяют в отгонах из вытяжек.
Данный метод является более чувствительным — чувствительность по аммиаку 0,06 мг/л.
а) Определение аммиака в водной вытяжке в присутствии акрилонитрила без омыления его. Для определения аммиака 3,5 мл вытяжки вносят в пробирку, добавляют в нее 0,5 мл 0,7-н серной кислоты, 1 мл 40% щелочи и 0,5 мл реактива Несслера. Смесь тщательно перемешивают и через 10 минут колориметрируют на фотоэлектроколориметре ФЭК-Н-57 с синим светофильтром N 3 (лямбда = 453 ммк) в кюветах с расстоянием между гранями 1 см.
Содержание аммиака в неомыленной вытяжке находят по графику зависимости D = f(C) (рис. 1 <*>, кривая 1).
<*> Рисунки не приводятся.
б) Определение общего количества аммиака: аммиака, содержащегося в исследуемой вытяжке, и аммиака, образовавшегося при омылении акрилонитрила. 3,5 мл вытяжки вносят в пробирку, добавляют 0,5 мл 0,7-н раствора серной кислоты, 1 мл 40% щелочи, закрывают пробирку воздушным холодильником на нормальном шлифе и помещают в кипящую водяную баню на 20 минут для омыления. После охлаждения раствора холодильник снимают, добавляют к раствору 0,5 мл реактива Несслера и через 10 минут колориметрируют на приборе ФЭК-Н-57.
Содержание аммиака в омыленной вытяжке также определяют по графику зависимости D = f(C) (рис. 1, кривая 1).
Содержание акрилонитрила в мг/л (А) определяют по формуле:
A = X x 3,12,
где X — разность содержаний аммиака в омыленной и неомыленной вытяжке, в мг/л; 3,12 — коэффициент пересчета аммиака на акрилонитрил.
Приготовление стандартной шкалы. Стандартные растворы аммиака и акрилонитрила (табл. 3) обрабатывают в той же последовательности, что и вытяжки.
Таблица 3
СТАНДАРТНАЯ ШКАЛА ДЛЯ ОПРЕДЕЛЕНИЯ АММИАКА И АКРИЛОНИТРИЛА В ВЫТЯЖКАХ
| Реактив | Номер стандарта | ||||||
| 0 | 1 | 2 | 3 | 4 | 5 | 6 | |
| Рабочий стандартный раствор, содержащий 10 мкг/мл аммиака или 31,2 мкг/мл акрилонитрила, мл | 0 | 0,1 | 0,2 | 0,4 | 0,6 | 0,8 | 1,0 |
| Серная кислота, 0,1-н раствор, мл | 3,5 | 3,4 | 3,3 | 3,1 | 2,9 | 2,7 | 2,5 |
| Едкое кали, 40% раствор | Во все пробирки по 1 мл | ||||||
| Реактив Несслера | Во все пробирки по 0,5 мл | ||||||
| Содержание аммиака, мкг | 0 | 1 | 2 | 4 | 6 | 8 | 10 |
| Содержание акрилонитрила, мкг | 0 | 3,12 | 6,26 | 12,48 | 18,72 | 24,96 | 31,2 |
По данным колориметрирования стандартных растворов строят график зависимости оптической плотности от концентрации аммиака при омылении акрилонитрила D = f(C) (см. рис. 1, кривая 1).
Определение аммиака и акрилонитрила в отгонах из вытяжек. 50 мл водной (или 2% уксуснокислой, или 5% солевой) (NaCl) вытяжки помещают в колбу Вюрца и отгоняют 9,5 мл раствора в градуированную пробирку с притертой пробкой, содержащую 1 мл 1-н серной кислоты (общий объем раствора 10,5 мл).
Для определения аммиака и акрилонитрила отбирают в две пробирки по 3,5 мл дистиллята и добавляют в них по 1 мл 40% щелочи; определение аммиака проводят без омыления и с омылением акрилонитрила, как указано в пунктах «а» и «б».
Содержание аммиака в неомыленном и омыленном отгоне определяют по графику зависимости D = f(C) (см. рис. 1, кривая 2).
Содержание акрилонитрила в мг/л (А) определяют по формуле:
где X — разность содержаний аммиака в омыленном и неомыленном отгонах, в мг/л; 3,12 — коэффициент пересчета аммиака на акрилонитрил.
Приготовление стандартной шкалы. Стандартные растворы аммиака и акрилонитрила приготовляют по данным табл. 4. Каждый стандартный раствор отгоняют в тех же условиях, что и вытяжку. Учитывая, что аммиак и акрилонитрил из 50 мл раствора могут быть количественно отогнаны только при условии отбора не менее 9,0 мл отгона, а анализу подвергается лишь 1/3 часть отгона, в 50 мл каждого стандартного раствора вносят утроенное количество аммиака или акрилонитрила.
При получении стандартной шкалы для определения аммиака из стандартного раствора хлорида аммония в каждые 50 мл приготовленного для отгона раствора следует добавить по 0,5 мл 40% щелочи, так как хлорид аммония — соль слабого основания и сильной кислоты. В данном случае отгоняют только 9,0 мл в 1,5 мл 1-н серной кислоты.
По данным колориметрирования стандартных растворов, приготовленных по табл. 4, строят график зависимости оптической плотности от концентрации аммиака (рис. 1, кривая 2).
Таблица 4
СТАНДАРТНАЯ ШКАЛА ДЛЯ ОПРЕДЕЛЕНИЯ АММИАКА И АКРИЛОНИТРИЛА В ОТГОНАХ ИЗ ВЫТЯЖЕК
| Реактив | Номер стандарта | ||||||
| 0 | 1 | 2 | 3 | 4 | 5 | 6 | |
| Рабочий стандартный раствор, содержащий 10 мкг/мл аммиака или 31,2 мкг/мл акрилонитрила, мл | 0 | 0,3 | 0,6 | 1,2 | 1,8 | 2,4 | 3,0 |
| Дистиллированная безаммиачная вода, мл | 50,0 | 49,7 | 49,4 | 48,8 | 48,2 | 47,8 | 47,0 |
| Дистилляция стандартных растворов | |||||||
| Общее количество дистиллята, мл | 10,5 мл в каждой пробирке | ||||||
| Количество дистиллята, взятое для определения, мл | 3,5 мл из каждой пробирки | ||||||
| Едкое кали, 40% раствор | Во все пробирки по 1 мл | ||||||
| Реактив Несслера | Во все пробирки по 0,5 мл | ||||||
| Содержание аммиака, мкг | 0 | 1 | 2 | 4 | 6 | 8 | 10 |
| Содержание акрилонитрила, мкг | 0 | 3,12 | 6,24 | 12,48 | 18,72 | 24,96 | 32,2 |
Примечание. Для определения акрилонитрила из 50 мл стандартного раствора отгоняют 9,5 мл дистиллята в 1 мл 1-н серной кислоты (общий объем пробы 10,5 мл). Для определения аммиака из 50 мл стандартного раствора отгоняют 9 мл дистиллята в 1,5 мл 1-н серной кислоты (общий объем пробы 10,5 мл). Для определения аммиака и акрилонитрила отбирают из 10,5 мл по 3,5 мл пробы.
ОПРЕДЕЛЕНИЕ АМИДА ОЛЕИНОВОЙ КИСЛОТЫ В ВОДНОЙ И МОЛОЧНОКИСЛОЙ ВЫТЯЖКАХ
Определение олеил-амида, или амида олеиновой кислоты, основывается на гидролизе его соляной кислотой с последующим обнаружением хлорида аммония с реактивом Несслера.
Метод позволяет обнаружить 0,001 мг азота в колориметрируемом объеме. На определение влияет аммиак и азотсодержащие соединения.
Реактивы
1. Соляная кислота, концентрированная.
2. Едкий натрий, 2-н раствор.
3. Безаммиачная дистиллированная вода.
4. Серная кислота, 0,1-н раствор.
5. Реактив Несслера.
6. Стандартный раствор хлористого аммония.
Ход определения
200 мл вытяжки выпаривают в фарфоровой чашке на кипящей водяной бане досуха, затем прибавляют в чашку 2 мл концентрированной соляной кислоты и снова выпаривают. После этого остаток из чашки переносят в колбу перегонного аппарата, применяя для этого безаммиачную дистиллированную воду — 100 мл, добавляют несколько стеклянных шариков или стеклянных капилляров, запаянных с одного конца. В приемную колбу наливают 10 мл 0,1-н раствора серной кислоты и опускают в нее трубку, соединенную с холодильником. В колбу перегонного аппарата через воронку по стенке наливают осторожно 100 мл 2-н раствора едкого натра, закрывают пробкой с каплеуловителем и соединяют с холодильником. Далее содержимое реакционной колбы перемешивают круговыми движениями и начинают нагревать. После полной отгонки аммиака (проба на аммиак) холодильник и трубку с шариком промывают безаммиачной дистиллированной водой. Отгон с промывной водой доводят в мерной колбе до 100 мл и приступают к определению аммиачного азота колориметрически с реактивом Несслера на ФЭК-М.
В качестве рабочего стандартного раствора применяют раствор хлористого аммония, содержащий в 1 мл 0,01 мг азота.
Для количественного определения аммиачного азота в пробирку наливают определенное количество (мл) испытуемого раствора (в зависимости от содержания азота), прибавляют 1 каплю едкого натра или кали, 0,1 мл реактива Несслера, содержимое доводят до объема 10 мл безаммиачной дистиллированной водой и тщательно перемешивают. Через 5 — 10 минут определяют интенсивность окраски на ФЭК-М с синим светофильтром (лямбда макс = 410 ммк) и кюветами с рабочей длиной 10 мм.
Количество аммиачного азота вычисляют по калибровочной кривой, составляемой по стандартной шкале (табл. 5).
Таблица 5
ШКАЛА СТАНДАРТОВ
| Реактив | Номер пробирки | |||||
| 1 | 2 | 3 | 4 | 5 | 6 | |
| Стандартный раствор, мл | 0,10 | 0,20 | 0,40 | 0,60 | 0,80 | 1,00 |
| Содержание азота, мг | 0,001 | 0,002 | 0,004 | 0,006 | 0,008 | 0,01 |
Расчет производится в мг азота на 1 л вытяжки или в мг амида олеиновой кислоты.
Для пересчета на амид олеиновой кислоты следует количество азота умножить на коэффициент 20,09 (частное от деления (281,288 / 14)).
Необходимо ставить контрольный опыт с применяемыми реактивами в тех же условиях.
ОПРЕДЕЛЕНИЕ БЕНАЗОЛА (ТИНУВИНА P, CH 3457, 2-(2′-ОКСИ-5′-МЕТИЛФЕНИЛ)-БЕНЗОТРИАЗОЛА) СПЕКТРОФОТОМЕТРИЧЕСКИМ МЕТОДОМ
Метод основан на измерении оптической плотности гексанового раствора 2-(2′-окси-5′-метилфенил)-бензотриазола при длине волны макс. 342 ммк с последующим количественным определением его по градуировочному графику.
Чувствительность метода 5 гамма во взятом для исследования объеме вытяжки. Ошибка метода +/- 1 гамма при количествах 2-(2′-окси-5′-метилфенил)-бензотриазола 5 — 10 гамма.
Определению не мешают водорастворимые количества бутилстеарата, стеарата цинка, Н-лаурилмеркаптана, диоктилфталата, полиграда, воднорастворимые соединения каучука — крилена, стирола и других веществ, не поглощающих свет при длине волны 342 ммк.
Реактивы и аппаратура
1. Спектрофотометр СФ-4А или другой марки.
2. 2-(2′-окси-5′-метилфенил)-бензотриазол, перекристаллизованный из спирта. Получают спиртовый раствор при нагревании до 60 град., фильтруют и охлаждают. Выпавшие кристаллы отфильтровывают и высушивают между листами фильтровальной бумаги.
3. Спирт этиловый 96 град., перегнанный.
4. Стандартный раствор 2-(2′-окси-5′-метилфенил)-бензотриазола в спирте, 1 мл = 50 гамма.
5. Н-гексан.
Построение калибровочного графика
Для построения калибровочного графика готовят:
1) гексановые экстракты из водных растворов, содержащих известное количество 2-(2′-окси-5′-метилфенил)-бензотриазола. Для этого в ряд делительных воронок вносят по 50 мл бидистиллированной воды и добавляют стандартный спиртовый раствор 2-(2′-окси-5′-метилфенил)-бензотриазола в количествах: 0,1; 0,2; 0,4; 0,6; 0,8 и 1,0 мл, смывают стенки каждой воронки 50 мл бидистиллированной воды, добавляют точно 6 мл н-гексана и извлекают 2-(2′-окси-5′-метилфенил)-бензотриазол путем осторожного экстрагирования в течение 3 минут, затем добавляют еще точно 4 мл н-гексана и снова осторожно экстрагируют в течение 3 минут. После разделения жидкостей сливают нижний водный слой как ненужный, а верхний слой — гексановую вытяжку — осторожно сливают через верхний край воронки в сухую пробирку с притертой пробкой. Во избежание попадания в гексановую вытяжку воды следует сливать ее не до конца;
2) одновременно готовят эталоны сравнения, не содержащие 2-(2′-окси-5′-метилфенил)-бензотриазола. Для этого в 6 делительных воронок вносят по 100 мл бидистиллированной воды и добавляют спирт в количестве, соответствующем содержанию его во взятом стандартном растворе для приготовления гексановых вытяжек с известными количествами 2-(2′-окси-5′-метилфенил)-бензотриазола, т.е. 0,1; 0,2; 0,4; 0,5; 0,8 и 1,0 мл; далее содержимое каждой воронки обрабатывают н-гексаном, как указано выше. Полученные гексановые вытяжки служат эталонами при определении оптической плотности в гексановых вытяжках, содержащих известные количества 2-(2′-окси-5′-метилфенил)-бензотриазола.
Оптическую плотность измеряют при лямбда макс = 342 ммк в кювете с толщиной слоя 1 см. Оптическая плотность соответствует количеству 2-(2′-окси-5′-метилфенил)-бензотриазола в 1 мл гексановой вытяжки, например 0,5 гамма/мл для вытяжки из первой делительной воронки.
По полученным данным строят калибровочный график, откладывая на оси абсцисс концентрацию 2-(2′-окси-5′-метилфенил)-бензотриазола в гамма/мл гексановой вытяжки, а на оси ординат — оптическую плотность.
Ход определения
100 мл вытяжки из исследуемого изделия помещают в делительную воронку, добавляют точно 6 мл н-гексана и извлекают 2-(2′-окси-5′-метилфенил)-бензотриазол путем осторожного взбалтывания в течение 3 минут; затем еще добавляют точно 4 мл н-гексана и снова взбалтывают в течение 3 минут.
После полного разделения жидкостей (обычно через 10 — 15 минут) нижний слой сливают как ненужный, а верхний — гексановую вытяжку — осторожно переносят через верхний край воронки в сухую пробирку с притертой пробкой (раствор N 1) <*>.
<*> Во избежание попадания в гексановую вытяжку воды ее сливают не до конца.
В другую воронку вносят 100 мл контрольного модельного раствора, примененного при получении вытяжки из изделия, и обрабатывают его 10 мл н-гексана аналогично исследуемой пробе. Гексановую вытяжку сливают в пробирку с притертой пробкой (раствор N 2). Каждый из растворов (N 1 и N 2) переносят в кювету с толщиной слоя 1 см и измеряют оптическую плотность раствора N 1 на спектрофотометре или лямбда макс = 342 ммк, при этом кювета с раствором N 2 служит эталоном.
Количество 2-(2′-окси-5′-метилфенил)-бензотриазола, отвечающее найденной оптической плотности, находят по калибровочному графику.
Количество 2-(2′-окси-5′-метилфенил)-бензотриазола в вытяжке из исследуемого изделия в мг в пересчете на 1 л (X) высчитывают по формуле:
| X = | C x а x 100 | = | C x a | , |
| V x 100 | V |
где C — количество 2-(2′-окси-5′-метилфенил)-бензотриазола в гамма/мл гексановой вытяжки, найденное по калибровочному графику; V — объем вытяжки, взятой для определения, в мл; а — количество н-гексана, взятое для извлечения 2-(2′-окси-5′-метилфенил)-бензотриазола, мл.
РЕАКЦИЯ НА ГАЛОИДЫ ПО БЕЛЬШТЕЙНУ
Реакция основана на способности раскаленной меди давать с галоидами летучие галоидные соединения меди, которые окрашивают пламя в зеленый цвет.
Реакцию дают: хлор, хлористый водород, бром, йод, галоидсодержащие органические вещества.
Реакция является высокочувствительной, однако некоторые, не содержащие галоидов, кислоты и азотсодержащие вещества (например, мочевина и некоторые производные пиридина) тоже могут давать летучие соединения меди, а следовательно, и положительную реакцию на галоиды.
Ход определения
Небольшую петлю на конце медной проволоки прокаливают на бунзеновской горелке до исчезновения зеленого окрашивания пламени, обусловленного наличием летучих солей меди, поверхность медной проволоки при этом покрывается окисью меди. После охлаждения петлю погружают в исследуемый раствор и вновь нагревают ее на пламени бунзеновской горелки. При наличии в растворе галоидов образуется галогенид меди, который улетучивается и окрашивает пламя в зеленый цвет. Необходимо проверить контрольный раствор на отсутствие в нем галоидсодержащих веществ.
ОПРЕДЕЛЕНИЕ ГЕКСАМЕТИЛЕНДИАМИНА В ВЫТЯЖКАХ
Метод основан на реакции взаимодействия гексаметилендиамина с 2,4-динитрохлорбензолом, при которой образуется 1,6-бис-(2,4-динитрофенил)-аминогексан желтого цвета.
Метод позволяет обнаружить 0,0025 мг гексаметилендиамина в колориметрируемом объеме. На определение влияет аммиак при концентрации 0,05 мг/л и выше.
Реактивы
1. Динитрохлорбензол, 5% спиртовый раствор.
2. Натрий углекислый, 8% раствор.
3. Соляная кислота, 5% раствор.
4. Хлороформ (для наркоза).
5. Стандартный раствор гексаметилендиамина, 1 мл = 1 мг, готовят на безаммиачной дистиллированной воде. Рабочий стандартный раствор, содержащий 0,01 мг гексаметилендиамина в 1 мл, готовят из основного раствора соответствующим разведением дистиллированной водой. Стандартный раствор сохраняется в течение 2 недель.
Ход определения
В колориметрическую пробирку помещают 2 мл исследуемой вытяжки <*>. Одновременно готовят шкалу стандартов (табл. 6).
<*> Вытяжки для определения гексаметилендиамина должны быть приготовлены на безаммиачной воде.
Таблица 6
СТАНДАРТНАЯ ШКАЛА ДЛЯ ОПРЕДЕЛЕНИЯ ГЕКСАМЕТИЛЕНДИАМИНА
| Реактив | Номер пробирки | ||||
| 0 | 1 | 2 | 3 | 4 | |
| Стандартный раствор, мл | 0 | 0,25 | 0,50 | 0,75 | 1,0 |
| Дистиллированная вода, мл | 2,0 | 1,75 | 1,50 | 1,25 | 1,0 |
| Содержание гексаметилендиамина, мл | 0 | 0,0025 | 0,005 | 0,0075 | 0,01 |
Примечание. Ввиду большой токсичности динитрохлорбензола и его сильного раздражающего действия на кожу и слизистые оболочки работу следует проводить с особыми предосторожностями, в резиновых перчатках и под тягой.
В пробирки с исследуемой вытяжкой и стандартными растворами добавляют 8% раствор углекислого натра до щелочной реакции раствора и избыток его в количестве 0,1 мл. Далее добавляют по 0,2 мл 5% раствора динитрохлорбензола. Содержимое пробирок перемешивают и ставят на 5 минут в кипящую водяную баню. После охлаждения до комнатной температуры в каждую пробирку добавляют 0,5 мл 5% раствора соляной кислоты и 1 мл хлороформа, после чего содержимое энергично встряхивают.
В присутствии гексаметилендиамина слой хлороформа окрашивается в зеленовато-желтый цвет. Интенсивность окраски зависит от концентрации гексаметилендиамина в растворе. Сравнивают окраску в пробирке с исследуемой вытяжкой со стандартной шкалой.
ОПРЕДЕЛЕНИЕ ГЕКСАМЕТИЛЕНДИАМИНА В МОЛОКЕ
Метод основан на выделении гексаметилендиамина посредством бумажной хроматографии с последующим качественным и количественным определением его по реакции с нингидрином.
Метод позволяет обнаружить 1 мкг в 1 мл колориметрируемого раствора, или 1 мг/л.
Реактивы
1. Уксусная кислота, 10% раствор.
2. Уксуснокислый свинец, 10% раствор.
3. Сернокислый натрий, 10% раствор.
4. Этиловый спирт, подкисленный соляной кислотой (100 мл этилового спирта и 0,5 мл конц. соляной кислоты).
5. Подвижный растворитель (н-пропанол, гидрат окиси аммония, вода 6:3:1).
6. Раствор нингидрина в подкисленном уксусной кислотой бутаноле (0,3 г нингидрина, 95 мл бутанола и 5 мл 2М раствора уксусной кислоты).
7. Стандартный раствор гексаметилендиамина (1 мл = 1 мг) готовят на дистиллированной воде; рабочий стандартный раствор, содержащий 0,01 мг гексаметилендиамина в 1 мл, готовят из основного раствора с соответствующим разведением дистиллированной водой. Стандартные растворы сохраняются в течение 2 недель.
Аппаратура и материалы
1. Аппарат для хроматографии: два цилиндрических сосуда с пришлифованными краями (диаметр — 20 см и высота аппарата — 50 см).
2. Кристаллизатор (диаметр — 50 см).
3. Бумага для хроматографического анализа марки «Б» ленинградской фабрики N 2 им. В. Володарского.
Ход определения
В один цилиндр емкостью 500 мл вносят 200 мл исследуемого молока, в другой — 200 мл контрольного молока, к которому добавляют стандартный раствор, содержащий гексаметилендиамин в количестве 0,3 мг («свидетель»), в оба цилиндра приливают по 20 мл 10% уксусной кислоты, по 50 мл 10% уксуснокислого свинца для осаждения белков и осторожно перемешивают стеклянной палочкой. После коагуляции осадка по капле прозрачного раствора из каждого цилиндра переносят на часовые стекла и добавляют к ним по капле уксуснокислого свинца; отсутствие мути указывает на полноту осаждения белков.
Объем содержимого каждого цилиндра доводят до 300 мл водой, перемешивают осторожно стеклянной палочкой и после отстаивания основной массы белков растворы фильтруют в конические колбы.
По 200 мл фильтрата переносят в мерные колбы емкостью 250 мл каждая, избыток уксуснокислого свинца удаляют из растворов, добавляя по каплям 10% раствор сернокислого натрия. Отсутствие мути от последующей капли сернокислого натрия указывает на полноту осаждения уксуснокислого свинца. Далее объем каждой колбы доводят до 250 мл, перемешивают и фильтруют. По 200 мл фильтрата из каждой колбы переносят небольшими порциями в две фарфоровые чашки, в которые предварительно вносят по 30 мл концентрированной соляной кислоты, и выпаривают на водяной бане досуха. Полученные остатки от исследуемого молока и «свидетеля» обрабатывают каждый 10 мл этилового спирта, подкисленного соляной кислотой.
Обработка остатков производится при помешивании стеклянной палочкой и нагревании на водяной бане при 60 град. C в течение 5 минут.
Спиртовые растворы сливают в цилиндры емкостью 25 мл, остатки в чашках еще раз обрабатывают 10 мл этилового спирта, подкисленного соляной кислотой, как указано выше, и присоединяют к первым извлечениям, доводя объемы растворов подкисленным этиловым спиртом до 20 мл. Затем растворы выдерживают в холодильнике в течение 2 часов. После этого их фильтруют и 15 мл каждого фильтрата выпаривают досуха в фарфоровых чашках на водяной бане. Каждый из остатков растворяют в 3 мл воды.
На определение берут по 0,1 мл исследуемого раствора и «свидетеля». Определение ведут из двух параллельных проб.
Для определения применяют лист хроматографической бумаги (45 x 30 см), на котором и отмечают стартовую линию (3 см от края). Исследуемые растворы (по 0,01 мл) наносят микропипеткой; нанесение новой порции раствора производят только после высыхания предыдущей капли.
Параллельные пробы наносят на бумагу на расстоянии 5 см друг от друга. По окончании нанесения проб лист бумаги свертывают в виде цилиндра и края сшивают белой ниткой так, чтобы они не накладывались один на другой.
В цилиндрический сосуд для хроматографирования наливают 150 — 200 мл подвижного растворителя и опускают в него подготовленный в виде цилиндра лист бумаги с нанесенными пробами (при этом бумага не должна касаться стенок сосуда), после чего его накрывают другим таким же цилиндрическим сосудом.
Хроматографирование проводят при температуре 25 — 26 град. в течение 6 часов. Далее лист бумаги вынимают из сосуда, сушат под тягой сначала на воздухе, а затем в сушильном шкафу при 105 град. C в течение 20 минут.
Высушенный лист хроматографической бумаги смачивают раствором нингидрина в подкисленном бутаноле путем протягивания его через раствор, налитый в кристаллизатор, после чего его высушивают под тягой. Для появления пятен хроматограммы выдерживают в термостате в течение 10 часов при температуре 25 — 26 град. C. При наличии гексаметилендиамина на хроматограмме появляются пятна сине-фиолетового цвета. Для гексаметилендиамина Rf = 0,8.
Проявленные хроматограммы подвергают качественному и количественному анализам.
Качественный анализ заключается в сравнении Rf сине-фиолетовых пятен, полученных из исследуемого образца молока, с Rf пятен, полученных из «раствора-свидетеля».
Для количественного определения гексаметилендиамина пятна из исследуемого молока, окрашенные в сине-фиолетовый цвет, вырезают из хроматограмм, помещают их в пробирки с притертыми пробками, добавляют по 3 мл воды, энергично встряхивают содержимое пробирок до обесцвечивания бумаги, фильтруют и определяют оптическую плотность фильтратов на фотоэлектроколориметре в кюветах с рабочей длиной 5 мм, зеленым светофильтром лямбда макс = 530 ммк. Эталоном служит вода.
Построение градуировочного графика
В ряд цилиндров на 500 мл каждый вносят стандартный раствор, содержащий гексаметилендиамин в количестве 0,03; 0,06; 0,12; 0,24; 0,3 мг, добавляют по 200 мл молока и обрабатывают точно так же, как описано для исследуемого молока.
Полученные пятна для каждой концентрации обрабатывают 3 мл воды и измеряют оптическую плотность полученных растворов.
На основании полученных данных строят градуировочный график, откладывая по оси абсцисс концентрацию гексаметилендиамина в мкг/мл, а по оси ординат — соответствующие величины оптической плотности.
По величине оптической плотности на градуировочном графике находят количество гексаметилендиамина в мкг/мл водного раствора. Количество гексаметилендиамина в мг (X) вычисляют по формуле:
| X = | а x б x 3 x 20 x 250 x 300 x 1000 | = | а x б x 300 | , |
| 0,1 x 15 x 200 x 200 x 200 | 0,8 |
где а — содержание гексаметилендиамина, найденное по градуировочному графику в 0,1 мл водного раствора пятна, в г; б — количество воды, взятое для обработки пятна, в мл.
ОПРЕДЕЛЕНИЕ ДИБУТИЛФТАЛАТА И ДИОКТИЛФТАЛАТА В ВОДНОЙ И NaCl-ВЫТЯЖКАХ
Метод основан на извлечении фталатов из вытяжек серным эфиром, их гидролизе с последующим определением продукта гидролиза — фталевой кислоты по реакции образования фенолфталеина в результате конденсации фталевого ангидрида с фенолом.
Реактивы
1. Серный эфир.
2. Фенол. К 100 г расплавленного на водяной бане фенола приливают при помешивании 10 г воды.
3. Серная кислота, концентрированная.
4. Спирт этиловый.
5. Натрий или калий едкий, 50% раствор.
6. Глицерин.
Ход определения
Для извлечения фталатов в делительную воронку емкостью 250 мл примешивают 50 — 100 мл испытуемой вытяжки, прибавляют 10 мл серного эфира и экстрагируют фталаты путем многократного перевертывания воронки в течение 5 минут. Водный слой сливают в другую делительную воронку, захватывая немного эфира. Экстракцию повторяют еще раз 10 мл эфира. Эфирные вытяжки сливают в фарфоровую чашечку и дают эфиру самопроизвольно испариться при комнатной температуре до объема 1 — 2 мл. Затем содержимое чашечки количественно переносят в тигель, смывая остаток в чашечке небольшим количеством эфира, и дают последнему полностью испариться при комнатной температуре (под тягой). После полного удаления эфира (отсутствие запаха) в тигель добавляют около 10 мг (одну каплю) фенола (реактив 2) и 1 — 2 капли концентрированной серной кислоты. Тигель помещают в глицериновую баню, нагретую до 100 град., и нагревают ее до 130 град. При этом происходит конденсация фталевого ангидрида с фенолом, содержимое тигля окрашивается в темно-красный (вишневый) цвет. Важно следить за тем, чтобы цвет плава не перешел в коричневый.
Далее тигель вынимают из бани, дают охладиться, растворяют плав в 1 мл спирта, добавляют 1 мл воды и затем по каплям щелочь до слабощелочной реакции. Рекомендуется, добавив первую каплю одной щелочи и тщательно смешав ее с раствором, подождать 1 минуту и лишь после этого проверить реакцию на лакмус. Последующие капли добавлять так же.
При наличии фталатов, в зависимости от их количества, раствор окрашивается в розовый цвет различной интенсивности.
Необходимо ставить контрольный опыт в аналогичных условиях с теми же реактивами.
Чувствительность метода около 0,2 мг в определенном объеме, или 2 мг/л.
ОПРЕДЕЛЕНИЕ ДИБУТИЛФТАЛАТА, ДИОКТИЛФТАЛАТА, БУТИЛСТЕАРАТА, ДИБУТИЛСЕБАЦИНАТА И АЦЕТИЛТРИБУТИЛЦИТРАТА КОЛОРИМЕТРИЧЕСКИМ МЕТОДОМ
Метод основан на гидролизе сложных эфиров серной кислотой и определении окрашенных продуктов взаимодействия полученных спиртов с ароматическими альдегидами.
Чувствительность метода 0,05 мг/л.
Определению мешают высшие спирты и эфиры. Стирол не мешает определению.
Реактивы и аппаратура
Хлороформ, х.ч. Хлороформ не должен окрашивать серную кислоту при встряхивании в делительной воронке.
Стандартный раствор соответствующего пластификатора в хлороформе, 0,10 и 0,01 мг/л.
Серная кислота (плотность 1,84).
М-нитробензальдегид (или П-диметиламинобензальдегид), 1% раствор в концентрированной серной кислоте.
Фотоэлектроколориметр ФЭК-Н-57.
Построение градуировочного графика
Для построения градуировочного графика готовят несколько стандартных шкал с содержанием вещества 0,005; 0,01; 0,03; 0,05; 0,07; 0,10 мг в зависимости от содержания пластификаторов в исследуемой вытяжке. Определенное количество стандартного раствора пластификатора в хлороформе вносят в делительную воронку со 100 мл модельного раствора и содержимое встряхивают в течение 1 минуты. Затем добавляют в воронку 10 мл хлороформа и опять встряхивают в течение 1 минуты. Дают смеси расслоиться и нижний слой сливают в пробирку. Далее проводят те же операции, что и при обработке вытяжек (см. ход определения). Затем измеряют оптическую плотность окрашенных растворов с синим светофильтром N 3 (лямбда = 453 ммк) и строят график зависимости D = f(C), где C — концентрация пластификатора в мг/л.
Следует отметить, что при использовании как М-нитробензальдегида, так и П-диметиламинобензальдегида получаются хорошо различимые шкалы.
Ход определения
В делительной воронке встряхивают 100 мл вытяжки с 10 мл хлороформа в течение 1 минуты. Дают смеси расслоиться и нижний слой (хлороформный) сливают в пробирку. Затем хлороформ выпаривают досуха на водяной бане, температура которой поддерживается на уровне 75 — 80 град. C. В охлажденную пробирку вносят 2,5 мл концентрированной серной кислоты и 0,5 мл 1% раствора П-диметиламинобензальдегида (или М-нитробензальдегида). Нагревают содержимое пробирки на кипящей водяной бане в течение 15 минут. В присутствии сложных эфиров растворы приобретают оранжево-красную, розовато-красную или красновато-коричневую окраску (в зависимости от вида пластификатора и типа модельной среды). Одновременно с пробами проводят холостой опыт (контроль на реактивы).
Оптическую плотность окрашенных растворов по отношению к контрольной пробе измеряют на фотоэлектроколориметре ФЭК-Н-57 с синим светофильтром N 3 (лямбда = 453 ммк) в кюветах с рабочей длиной 3 мм. Концентрацию пластификатора находят по градуировочному графику D = f(С).
ОПРЕДЕЛЕНИЕ ДИБУТИЛСЕБАЦИНАТА, ДИБУТИЛ- И ДИОКТИЛФТАЛАТОВ В МАСЛЯНЫХ ВЫТЯЖКАХ МЕТОДОМ ГАЗОВОЙ ХРОМАТОГРАФИИ
Газохроматографический метод основан на различии коэффициентов распределения веществ между двумя фазами: газовой и жидкой, нанесенной на твердый носитель.
Метод высокотемпературной газовой хроматографии позволяет анализировать пластификаторы без предварительного их преобразования (омыление, метилирование), благодаря чему повышается точность анализа, сокращается число операций и продолжительность определения.
Чувствительность метода 5 — 10 мг пластификатора в 1 л масляной вытяжки в зависимости от физико-химических свойств пластификатора.
Реактивы и аппаратура
1. Газовый хроматограф с пламенно-ионизационным детектором (из отечественных хроматографов рекомендуются: Цвет-1, Цвет-2 и др., ЛХМ-7а; из импортных: Хром-2, Хром-31 и др.).
2. Жидкая фаза (апиезон, силиконовые каучуки марок SE-30 и E-52, резофлекс LAC-2R-446).
3. Твердый носитель (целит С-22, целит-545, хромосорбы W, R, G).
4. Газ-носитель (аргон, азот).
5. Водопровод электролизный.
6. Сжатый воздух.
7. Нитрометан, ч.д.а.
8. Внутренний стандарт (дибутилфталат (ДБФ), дибутилсебацинат (ДСБ), диаминовый эфир себациновой кислоты (ДАС), ч.д.а. или х.ч.).
Построение калибровочного графика
Количественное определение пластификатора проводят по внутренним стандартам. В качестве стандартного вещества для ДБС применяют ДБФ, для ДБФ — ДБС, для ДОФ — ДАС.
С целью построения градуировочных графиков готовят смеси пластификаторов и стандартных веществ в нитрометане с различным содержанием каждого из компонентов. Соотношение пластификатора и стандарта выбирается так, чтобы площади их пиков были примерно одинаковыми. По оси абсцисс откладывают известные количества пластификатора, а по оси ординат — его количества, найденные относительно внутреннего стандарта по формуле:
где Qx — количество пластификатора, найденное относительно внутреннего стандарта, мг; Sx — площадь пика пластификатора, кв. мм; Qc — количество добавленного внутреннего стандарта, мг; Sc — площадь пика внутреннего стандарта, кв. мм.
Площадь пика равна произведению его высоты на ширину, взятую на половине высоты пика.
Калибровочный график для определения содержания ДБС и ДОФ представлен на рис. 2 и 3.
Ход определения
В делительную воронку отбирают 10 — 20 мл исследуемой масляной вытяжки, добавляют 10 мл нитрометана и проводят извлечение пластификаторов путем осторожного перевертывания воронки (50 — 60 раз). Экстракцию повторяют 5 — 7 раз. Полученные экстракты сгущают под вакуумом до объема около 1 мл в конической перегонной колбе, а затем переносят в пробирку с притертой пробкой. Чтобы установить примерное содержание пластификатора в экстракте, с помощью микрошприца отбирают около 5 мкл пробы, вводят в хроматограф и проводят определение. Вычисляют площадь полученного пика и по калибровочному графику находят примерное содержание определяемого пластификатора. Исходя из полученных данных, прибавляют к экстракту внутренний стандарт в таком количестве, чтобы площади пиков искомого и добавляемого веществ были близки по величине.
Снова отбирают пробу около 5 мкл и хроматографируют. По величине обеих площадей и количеству добавленного стандарта вычисляют относительное содержание пластификатора, а затем по калибровочному графику находят фактическое содержание его в анализируемой вытяжке. Образцы хроматограмм экстрактов ДБС, ДБФ и ДОФ из масляных вытяжек с добавлением внутренних стандартов приведены на рис. 4, 5.
Для анализа пластификаторов рекомендуется применять короткие колонки из нержавеющей стали длиной 0,5 — 1 м, количество жидкой фазы 5 — 10% от веса твердого носителя. Рабочая температура 230 — 240 град. C. Расход газа-носителя — 50 мл/мин., водорода — 50 мл/мин., воздуха — 850 мл/мин.
ОПРЕДЕЛЕНИЕ ДИМЕТИЛОВОГО ЭФИРА ТЕРЕФТАЛЕВОЙ КИСЛОТЫ (ДИМЕТИЛТЕРЕФТАЛАТА)
Метод основан на омылении диметилтерефталата раствором щелочи и на последующем определении образующегося в результате его гидролиза метилового спирта после окисления его до формальдегида по реакции с хромотроповой кислотой.
Метод позволяет обнаружить 0,01 мг или 10 гамма диметилтерефталата в колориметрируемом объеме. Метод неспецифичен, определению мешают формальдегид, метиловый спирт и сложные эфиры, содержащие метильную группу.
Реактивы
Едкий натр, 5% водный раствор.
Ход определения
50 мл испытуемой вытяжки помещают в колбу для омыления и прибавляют половинный объем 5% раствора щелочи. Присоединяют колбу к шариковому холодильнику и погружают ее на 30 минут в водяную баню (температура 80 — 90 град.) для гидролиза диметилтерефталата.
Одновременно ставят контрольный опыт с реактивами.
Содержимое колбы после омыления переносят в колбу перегонного аппарата, смывая остатки с колбы небольшим количеством дистиллированной воды (5 мл) и отгоняют 40 мл дистиллята; приемник должен быть погружен в воду со льдом. В дистилляте определяют метиловый спирт после окисления его до формальдегида по реакции с хромотроповой кислотой.
При наличии метилового спирта в вытяжках содержание диметилтерефталата определяют по разности между общим количеством метилового спирта, образовавшегося в результате гидролиза в щелочной среде, и количеством метилового спирта до гидролиза.
Содержание диметилтерелфталата (X) в мг/л рассчитывают по следующей формуле:
| X = | 0,01 x a x 40 x 1000 x 3,0 | , |
| 2 x г |
где а — количество рабочего стандартного раствора метилового спирта, интенсивность окраски которого соответствует исследуемой вытяжке, в мл; г — объем вытяжки, взятой для исследования, в мл; 3,0 — коэффициент пересчета метилового спирта на диметилтерефталат; 2 — количество дистиллята, взятое для определения, в мл; 40 — общее количество дистиллята, в мл.
ОПРЕДЕЛЕНИЕ ДИФЕНИЛОЛПРОПАНА
Метод основан на получении нитрозосоединения при взаимодействии дифенилолпропана с азотистой кислотой и колориметрическом определении образующегося в щелочной среде продукта реакции желтого цвета.
Метод позволяет обнаружить 0,002 мкг (2 гамма) дифенилолпропана в колориметрируемом объеме, или 0,02 мг/л.
Эпихлоргидрин, толуол не мешают определению.
Реактивы
1. Едкий натр, 20% раствор.
2. Едкий натр, 0,1-н раствор.
3. Серная кислота, 20% раствор (по весу).
4. Азотистокислый натрий, 0,5% раствор, свежеприготовленный.
5. Аммиак, 15% раствор.
6. Эфир серный, обезвоженный сульфатом натрия и перегнанный на водяной бане (с температурой 40 — 50 град.), остаток при перегонке, примерно около 10% от взятого объема, отбрасывают.
7. Стандартный раствор дифенилолпропана.
Готовят основной раствор: 0,01 г дифенилолпропана растворяют в мерной колбе емкостью 100 мл в 0,1-н растворе NaOH, в случае необходимости — при подогревании в водяной бане. После охлаждения раствор доводят до метки 0,1-н раствором щелочи (1 мл = 0,1 мг дифенилолпропана).
Для приготовления рабочего стандартного раствора 10 мл основного раствора переносят в мерную колбу на 100 мл и доводят до метки 0,1-н раствором NaOH (1 мл = 0,01 мг дифенилолпропана). Раствор готовят в день определения.
Ход определения
100 мл исследуемой вытяжки (водной, уксуснокислой, виннокислой, а также вытяжки, полученной при обработке изделия поваренной солью) переносят в делительную воронку и без подщелачивания экстрагируют 20 мл эфира путем энергичного перевертывания воронки 40 — 50 раз, не допуская взбалтывания раствора во избежание образования стойкой эмульсии.
При исследовании молочнокислой вытяжки последнюю предварительно нейтрализуют 20% раствором едкого натра, пользуясь при этом универсальной индикаторной бумагой, после чего проводят экстракцию дифенилолпропана 20 мл эфира, как указано выше.
При определении дифенилолпропана в спиртовых вытяжках или вытяжках, содержащих спирт, спирт предварительно удаляют на водяной бане.
После расслоения жидкости водный слой сливают через кран в другую делительную воронку, в которой повторяют экстракцию 10 мл эфира. Соединенные эфирные вытяжки испаряют в фарфоровой чашке при комнатной температуре в вытяжном шкафу.
Остаток в чашке количественно переносят с помощью 5 мл 0,1-н раствора едкого натра в колориметрическую пробирку с притертой пробкой, беря каждый раз по 1 мл. Объем раствора в пробирке доводят 0,1-н едким натром до 5 мл.
К 5,0 мл полученного раствора в колориметрической пробирке с притертой пробкой приливают 0,5 мл 20% раствора H2SO4 и 0,5 мл раствора NaNO3. Содержимое пробирки нагревают в течение 5 минут в кипящей водяной бане. По охлаждении к раствору добавляют 2 мл 15% раствора аммиака. Через 30 минут сравнивают интенсивность желтой окраски со стандартной шкалой, одновременно приготовленной в тех же условиях, или измеряют оптическую плотность раствора на фотоэлектроколориметре ФЭК-Н-57 при длине волны 413 ммк в кювете с толщиной слоя 20 мм. Количество дифенилолпропана, отвечающее найденной оптической плотности, находят по калибровочному графику, для построения которого измеряют оптические плотности растворов той же стандартной шкалы (табл. 7).
Таблица 7
СТАНДАРТНАЯ ШКАЛА ДЛЯ ОПРЕДЕЛЕНИЯ ДИФЕНИЛОЛПРОПАНА
| Реактив | Номер пробирки | |||||
| 1 | 2 | 3 | 4 | 5 | 6 | |
| Стандартный раствор дифенилолпропана (1 мл = 0,01 мг), мл | 0 | 0,20 | 0,40 | 0,60 | 0,80 | 1,00 |
| NaOH, 0,1-н, мл | 5,0 | 4,8 | 4,6 | 4,4 | 4,2 | 4,0 |
| Содержание дифенилолпропана, мг | 0 | 0,002 | 0,004 | 0,006 | 0,008 | 0,01 |
При этом интенсивность образующейся желтой окраски пропорциональна концентрации дифенилолпропана в растворе.
ОПРЕДЕЛЕНИЕ КАНЦЕРОГЕННЫХ АРОМАТИЧЕСКИХ УГЛЕВОДОРОДОВ, В ТОМ ЧИСЛЕ 3,4-БЕНЗПИРЕНА
Определение люминесценции парафина при ультрафиолетовом облучении.
Реактивы и аппаратура
Аппарат для люминесцентного (флуоресцентного) анализа витаминов в растворах или любой другой аналогичный аппарат, где источником возбуждения ультрафиолетовых лучей служит ртутно-кварцевая лампа типа ПРК-4.
Светофильтр типа УФС-2 или УФС-3.
Насос стеклянный водоструйный лабораторный по ГОСТ 10696-63 или любой другой вакуумный насос.
Пробирки стеклянные по ГОСТ 10515-63, тип ПХ-14.
Стаканы стеклянные лабораторные по ГОСТ 10394-63, тип ПН, вместимостью 25 мл.
Меры вместимости стеклянные технические по ГОСТ 1770-64.
Колба мерная, тип 1, емкостью 1000 мл.
Колба мерная, тип 1, емкостью 100 мл.
Пипетка на 5 мл, тип 1, с делениями.
Кислота серная по ГОСТ 4204-66, х.ч. или ч.д.а, 0,1-н раствор.
3,4-бензпирен, эталонный раствор.
Н-октан, дважды перегнанный, нелюминесцирующий.
Азот жидкий технический по ГОСТ 9293-59.
Ход определения
Для определения люминесценции парафина в стаканчик емкостью 25 мл берут 1,0 г стружки парафина, снятой с трех различных мест образца, и экстрагируют 4 мл дважды перегнанного нелюминесцирующего н-октана при интенсивном перемешивании при комнатной температуре (но не выше 30 град.) в течение 30 минут.
Экстракт масел и ароматических углеводородов в растворе н-октана отделяют от выкристаллизовавшегося парафина декантацией или вакуумной фильтрацией в пробирку.
В другую пробирку помещают 1 мл эталонного раствора 3,4-бензпирена в концентрации 1,10(-10) г/мл и 3 мл дважды перегнанного н-октана. Обе пробирки помещают рядом в держателях пробирок в прозрачный сосуд Дьюара, наполненный жидким азотом.
Пробы облучают ультрафиолетовым светом, сфокусированным конденсором на пробирках, и сравнивают.
Образец парафина считают выдержавшим испытание, если люминесценция замороженного экстракта из парафина не видна по сравнению с люминесценцией замороженного н-октанового раствора 3,4-бензпирена.
Всю стеклянную посуду, используемую при испытании парафина, тщательно промывают хромовой смесью, затем дистиллированной водой и просушивают (ГОСТ 13577-68, парафин нефтяной для пищевой промышленности).
ОПРЕДЕЛЕНИЕ Е-КАПРОЛАКТАМА
Качественное определение
Реакция основана на взаимодействии Е-капролактама с тетрайодвисмутитом калия (KBiJ4) с образованием кристаллического осадка в виде красных и темно-красных кристаллов гексагональной системы следующего химического состава (C6H11NOH)2BiJ4. Чувствительность реакции для водных растворов 0,01 мг в определяемом объеме.
Реактивы
1. Раствор йодвисмутита калия: 5,825 г окиси висмута (Bi2O3) растворяют в 10 мл концентрированной соляной кислоты (уд. в. 1,19) и выпаривают в фарфоровой чашке на водяной бане досуха.
Полученный хлористый висмут растворяют в 50 мл дистиллированной воды. 50 г йодистого калия (ч.д.а.) растворяют в небольшом количестве дистиллированной воды, подкисляют 0,5 мл концентрированной соляной кислоты, оба раствора сливают вместе и доводят объем до 100 мл дистиллированной водой. Раствор должен храниться в склянке из темного стекла.
2. Азотная кислота, 5% раствор.
Ход определения
25 — 50 мл испытуемой вытяжки упаривают в фарфоровой чашке на водяной бане до небольшого объема (около 1 мл). В сконцентрированном растворе проводят качественную реакцию на Е-капролактам. На предметное стекло наносят 1 — 2 капли раствора и осторожно испаряют на теплой водяной бане.
К сухому остатку на предметном стекле прибавляют небольшую каплю 5% раствора азотной кислоты и каплю реактива йодвисмутита калия, не смешивая их, давая им натечь, или соединяют капли оплавленной стеклянной нитью. В присутствии Е-капролактама отмечается появление одиночных и сдвоенных кристаллов в виде шестиугольных призм, группирующихся также в пучки и цепочки красного и темно-красного цвета.
Количественное определение по Д.А. Бабаеву
Реакция основана на образовании гидроксамовой кислоты при взаимодействии Е-капролактама с сернокислым гидроксиламином в щелочной среде; гидроксамовая кислота с трехвалентным железом дает окрашенное в коричневый (при малых количествах капролактама) до темно-красно-фиолетового цвета (при больших количествах Е-капролактама) комплексное соединение.
Чувствительность метода 0,01 мг/мл.
Реактивы
1. 0,74-молярный раствор хлорного железа, приготовленный на 0,1-н растворе соляной кислоты.
2. Молярный раствор сернокислого гидроксиламина.
3. 4,5-н раствор соляной кислоты.
4. 4,5-н раствор едкого натра.
Ход определения
В пробирку с притертой пробкой емкостью 10 мл наливают 1 мл 4,5-н раствора едкого натра и 1 мл сернокислого гидроксиламина. К раствору прибавляют 1 — 2 мл испытуемой вытяжки. После тщательного перемешивания пробирку помещают в кипящую водяную баню на 30 минут. Затем раствор путем погружения пробирки в холодную воду быстро охлаждают до комнатной температуры. К нему добавляют 1 мл 4,5-н раствора соляной кислоты и 1 мл 0,74-молярного раствора хлорного железа. При наличии Е-капролактама раствор приобретает коричневый или красно-фиолетовый цвет. Одновременно в тех же условиях проводят контрольную пробу, для чего вместо вытяжки берут 1 — 2 мл дистиллированной воды. Контрольная проба светло-зеленоватого цвета.
Приготовленные растворы едкого натра и соляной кислоты, взятые в равных объемах, должны иметь нейтральную реакцию, иначе после добавления хлорного железа образуется бурый осадок, что указывает на щелочную реакцию раствора.
Количественное определение Е-капролактама и низкомолекулярных азотсодержащих соединений
Метод основан на гидролизе Е-капролактама и низкомолекулярных азотсодержащих соединений в присутствии серной кислоты с последующей минерализацией по методу Кьельдаля и определением аммонийного азота колориметрическим методом с реактивом Несслера.
Чувствительность метода 0,001 мг азота в колориметрируемом объеме.
Реактивы
Приготовление см. выше.
Ход определения
25 — 50 мл испытуемой вытяжки подкисляют серной кислотой до получения 10% концентрации ее в растворе и содержимое кипятят с обратным холодильником в течение 6 — 8 часов для гидролитического расщепления определяемых веществ. Гидролизат количественно переносят в колбу Кьельдаля емкостью 250 мл, добавляют 10 мл концентрированной серной кислоты (уд. в. 1,835 — 1,84), 2 — 3 капли 10% раствора сернокислой меди и 1 — 2 мл 10% сульфата калия и далее проводят работу, как указано выше.
Расчет производят в мг азота.
Коэффициент пересчета количества азота на Е-капролактам 8,08
| (частное от деления | 113,162 | ). |
| 14 |
ОПРЕДЕЛЕНИЕ МЕЛАМИНА
25 — 50 мл вытяжки выпаривают на водяной бане до небольшого объема (около 1 мл) и проводят указанные ниже реакции.
Микрореакция по Архангелову
Каплю раствора переносят на предметное стекло, прибавляют каплю раствора пикриновой кислоты и обе капли быстро перемешивают оплавленной стеклянной нитью, что способствует кристаллизации. При наличии меламина образуются игольчатые кристаллы, группирующиеся в пучки, снопики и звездчатые скопления.
Реактивы
Пикриновая кислота, 0,1% раствор.
Микрореакция по Саркисянц
Каплю исследуемого раствора переносят на предметное стекло и добавляют каплю реактива Драгендорфа. При наличии меламина образуются рубиново-красные игольчатые, ромбовидные, линзовидные кристаллы, частично группирующиеся в разнообразные скопления в виде звездочек, крестиков и т.п. Прибавление к исследуемой капле 1 — 2% раствора азотной кислоты способствует реакции.
Чувствительность 0,01 мг.
Реактивы
Реактив Драгендорфа: 8 г основного азотнокислого висмута растворяют в 20 г азотной кислоты (уд. в. 1,18) и вливают в концентрированный раствор из 27,2 г йодистого калия в 30 мл дистиллированной воды. Через несколько дней раствор отфильтровывают от выделившейся селитры, а фильтрат разбавляют дистиллированной водой до 100 мл.
ОПРЕДЕЛЕНИЕ МЕТИЛОВОГО СПИРТА
Определение метилового спирта с фуксиносернистой кислотой
Метод основан на окислении метилового спирта в кислой среде перманганатом калия до формальдегида с последующим проведением цветной реакции с фуксиносернистой кислотой.
Отметим, что фуксиносернистая кислота является общим реактивом для альдегидов, но в присутствии серной или соляной кислот обнаруживается только формальдегид.
Метод позволяет обнаружить содержание метилового спирта в количестве 0,05 мг в колориметрируемом объеме.
Реактивы
1. Серная кислота, концентрированная (уд. в. 1,835 — 1,84).
2. Перманганат калия, 5% раствор.
3. Щавелевая кислота, 8% раствор.
4. Смесь серной кислоты с этиловым спиртом. 2 мл спирта и 4 мл концентрированной серной кислоты доводят водой до 100 мл.
5. Раствор фуксиносернистой кислоты. 0,2 г химически чистого основного кристаллического фуксина растворяют в 120 мл бидистиллированной горячей воды. По охлаждении раствора добавляют 6 г безводного сульфита натрия, растворенного в 20 мл бидистиллированной воды, 4 мл соляной кислоты (уд. в. 1,19) и доводят бидистиллированной водой до 200 мл. Содержимое колбы фильтруют и помещают в темную склянку с притертой пробкой. Реактив должен быть бесцветным или слегка желтоватым.
6. Стандартный раствор метилового спирта (метанола). Готовят исходный раствор: в мерную колбу на 50 мл наливают 20 мл дважды перегнанной воды, затем колбу взвешивают на аналитических весах. Вносят в колбу 3 — 4 капли перегнанного метилового спирта и вновь взвешивают. По разности веса определяют количество метилового спирта. Раствор в колбе доводят до метки водой. Затем высчитывают содержание метилового спирта в 1 мл раствора.
Из этого раствора готовят стандартный раствор с содержанием метанола 10 мг в 1 мл. Перед определением готовят рабочий стандартный раствор с содержанием метилового спирта 0,1 мг в 1 мл.
Ход определения
200 мл испытуемой вытяжки помещают в колбу аппарата для перегонки с вертикально установленным небольшим холодильником и отгоняют приблизительно 100 мл. Полученный дистиллят отгоняют второй раз, собирая около 50 мл. Наконец, при третьем отгоне из 50 мл получают 20 мл конечного дистиллята (содержащего весь метиловый спирт исходной пробы). Таким образом, концентрация метилового спирта увеличивается в 10 раз по сравнению с исследуемой вытяжкой. Колбы — приемники дистиллята при отгоне должны быть погружены в воду со льдом.
К 3 мл дистиллята в колориметрической пробирке с притертой пробкой приливают 1 мл смеси спирта с серной кислотой и 1 мл 5% раствора перманганата калия. Смесь перемешивают, через 2 минуты прибавляют 1 мл 8% раствора щавелевой кислоты и вновь перемешивают легким взбалтыванием. Содержимое пробирки должно обесцветиться. Затем прибавляют 1 мл концентрированной серной кислоты и, после перемешивания, 5 мл фуксиносернистой кислоты, содержимое вновь перемешивают и оставляют стоять в течение часа.
В присутствии метилового спирта, окисленного перманганатом до формальдегида, появляется фиолетовое, а при малых его количествах — голубое окрашивание. Параллельно необходимо ставить контрольный опыт с реактивами при тех же условиях.
Для количественного определения метилового спирта в дистилляте одновременно проводят реакцию с фуксиносернистой кислотой со стандартными растворами и с дистиллятом.
Готовят шкалу, как указано в табл. 8.
Таблица 8
| Реактив | Номер пробирки | ||||||
| 0 | 1 | 2 | 3 | 4 | 5 | 6 | |
| Стандартный раствор (1 мл = 0,1 мг метилового спирта), в мл | 0 | 0,50 | 1,0 | 1,50 | 2,0 | 2,50 | 3,0 |
| Дистиллированная вода, мл | 3,0 | 2,50 | 2,0 | 1,50 | 1,0 | 0,50 | 0 |
| Содержание метилового спирта, мг | 0 | 0,05 | 0,10 | 0,15 | 0,20 | 0,25 | 0,3 |
В ряд одинаковых колориметрических пробирок вводят определенное количество рабочего стандартного раствора метилового спирта (1 мл = 0,1 мг метилового спирта).
Во всех пробирках объем жидкости доводят дистиллированной водой до объема испытуемого раствора, взятого на определение.
Определение метилового спирта с хромотроповой кислотой
Метод основан на окислении метилового спирта в кислой среде перманганатом калия до формальдегида с последующим проведением цветной реакции с хромотроповой кислотой.
Метод позволяет обнаружить содержание метилового спирта в количестве 0,001 мг (или 1 гамма) в колориметрируемом объеме (0,25 мг/л).
Метод неспецифичен, присутствие формальдегида мешает определению.
Реактивы
1. Серная кислота, 25% раствор.
2. Серная кислота, концентрированная (уд. в. 1,835 — 1,84).
3. Перманганат калия, 0,2% раствор.
4. Сульфит натрия (Na2SO3 x 7H2O), 5% раствор.
5. Хромотроповая кислота (1,8-диоксинафталин-3, 6-дисульфокислота) или ее динатриевая соль (C10H6O8Na2), 1% водный раствор, свежеприготовленный.
6. Стандартный раствор метилового спирта. Готовят исходный раствор. В мерную колбу емкостью 50 мл наливают 20 мл дважды перегнанной воды, затем колбу взвешивают на аналитических весах. Вносят в колбу 3 — 4 капли перегнанного метилового спирта и вновь взвешивают. По разности веса определяют количество метилового спирта. Раствор в колбе доводят до метки бидистиллированной водой, затем высчитывают содержание метилового спирта в 1 мл раствора. Перед определением из исходного раствора готовят рабочий стандартный раствор с содержанием метилового спирта 0,01 мг, или 10 гамма/мл.
Ход определения
100 мл испытуемой вытяжки помещают в круглодонную колбу аппарата для перегонки с небольшим холодильником и осторожно отгоняют 50 мл в мерную колбу. Колба — приемник дистиллята при отгоне должна быть погружена в воду со льдом.
2 мл дистиллята переносят в колориметрическую пробирку с притертой пробкой, приливают 0,5 мл 25% раствора серной кислоты и 0,2 мл 0,2% раствора перманганата калия. Содержимое пробирки встряхивают (в течение 5 секунд) и оставляют стоять в течение 5 минут, после чего добавляют по каплям 5% раствор сульфита натрия до обесцвечивания (избегать избытка).
В пробирку с обесцвеченным раствором добавляют 0,4 мл 1% раствора хромотроповой кислоты или ее динатриевой соли и 1,7 мл концентрированной серной кислоты. Нагревают пробирку в кипящей водяной бане в течение 30 минут, по охлаждении содержимое пробирки перемешивают и оставляют стоять в течение 40 — 60 минут.
В присутствии метилового спирта, окисленного перманганатом до формальдегида, образуется красновато-фиолетовое, а при малых его концентрациях — розовое окрашивание жидкости.
Параллельно следует ставить контрольный опыт с реактивами при тех же условиях.
Для количественного определения метилового спирта в дистилляте одновременно проводят реакцию с хромотроповой кислотой со стандартными растворами и с дистиллятом. Шкалу готовят, как указано в табл. 9.
Таблица 9
СТАНДАРТНАЯ ШКАЛА ДЛЯ ОПРЕДЕЛЕНИЯ МЕТИЛОВОГО СПИРТА
| Реактив | Номер пробирки | ||||||
| 0 | 1 | 2 | 3 | 4 | 5 | 6 | |
| Стандартный раствор (1 мл = 0,01 мг метилового спирта), мл | 0 | 0,10 | 0,20 | 0,40 | 0,60 | 0,80 | 1,00 |
| Дистиллированная вода, мл | 2,0 | 1,90 | 1,80 | 1,60 | 1,40 | 1,20 | 1,00 |
| Содержание метилового спирта, гамма/мл | 0 | 1,0 | 2,0 | 4,0 | 6,0 | 8,0 | 10,0 |
| 0 | 0,001 | 0,002 | 0,004 | 0,006 | 0,008 | 0,010 |
В ряд одинаковых колориметрических пробирок вводят определенное количество рабочего стандартного раствора метилового спирта (1 мл = 0,01 мг метилового спирта).
Во всех пробирках объем жидкости доводят до объема дистиллята, взятого на определение.
ОПРЕДЕЛЕНИЕ МОЧЕВИНЫ В ВОДНОЙ ВЫТЯЖКЕ
Метод основан на минерализации мочевины с помощью серной кислоты с образованием сернокислого аммония.
Разложение сернокислого аммония щелочью, поглощение аммиака титрованным раствором серной кислоты с последующим определением связанного им количества серной кислоты проводится в чашках Конвея.
Чувствительность метода 0,01 мг мочевины в мл раствора, полученного после минерализации. Ошибка метода +/- 10%.
Метод неспецифичен, другие азотсодержащие соединения будут определяться вместе с мочевиной.
Реактивы и посуда
1. Серная кислота (уд. вес 1,84).
2. Натр едкий, 20% раствор.
3. Серная кислота, 0,01-н раствор.
4. Натр едкий, 0,01-н раствор.
5. Пергидроль.
6. Натрий углекислый, 10% раствор.
7. Безаммиачная дистиллированная вода. Безаммиачную воду получают вторичной перегонкой дистиллированной воды, подкисленной серной кислотой с добавлением перманганата калия.
8. Индикатор Ташира. Основной раствор: к 40 мл 0,1% спиртового раствора метилового красного добавляют 10 мл 0,1% спиртового раствора метиленового синего. Рабочий раствор: к 1 объему основного раствора добавляют 1 объем спирта и 2 объема воды. Цвет раствора зеленый — в щелочной среде и винно-красный — в кислой.
9. Смазка для чашек Конвея: в фарфоровую чашку вносят 300 г вазелина (технического), 50 г воска (х.ч.) и 50 г парафина. Чашку ставят на водяную баню до расплавления (осторожно с огнем — пары парафина дают вспышку!), хорошо перемешивают до получения однородной массы, охлаждают и помещают в банку с притертой пробкой.
10. Чашки Конвея. Чашки по форме напоминают кристаллизатор с низкими стенками, в центральную часть которого впаян цилиндр. Кроме того, наружная камера чашки разделена стеклянной перегородкой. Высота чашки 1,5 см, высота цилиндра 1 см. Диаметр чашки равен 7 — 8 см, диаметр цилиндра — 3 см. Чашки плотно закрывают пришлифованными стеклянными крышками.
Ход определения
50 мл исследуемой вытяжки переносят в фарфоровую чашку, добавляют углекислый натрий до слабощелочной реакции и выпаривают на водяной бане до объема около 5 мл. Остаток переносят в колбочку Кьельдаля объемом 25 мл путем 5-кратного споласкивания фарфоровой чашки безаммиачной дистиллированной водой, беря по 1 мл каждый раз; добавляют 1 мл концентрированной серной кислоты и проводят минерализацию на песчаной бане до полного обесцвечивания раствора. Если раствор не обесцвечивается, а тяжелые пары серной кислоты уже появились, то в колбочку добавляют 10 капель пергидроля и кипятят еще 15 — 20 минут.
Далее бесцветное содержимое колбочки Кьельдаля после охлаждения количественно переносят в мерную колбу на 25 мл и доводят с помощью безаммиачной дистиллированной воды содержимое колбы до метки.
В полученном растворе определяют аммонийный азот методом Конвея. Перед заполнением чашки Конвея шлиф чашки хорошо смазывают смазкой. После этого во внутреннюю камеру чашки наливают 2 мл 0, 01-н раствора серной кислоты и 5 капель индикатора Ташира, чашку ставят немного наклонно и во внешнюю камеру с одной стороны перегородки наливают 1 мл испытуемого раствора, не давая раствору растекаться по всей поверхности чашки; с другой стороны перегородки наливают 3 мл 20% раствора едкого натра. Тотчас закрывают чашку крышкой, плотно притирая ее к краям чашки, ставят в горизонтальное положение и вращательным движением осторожно перемешивают содержимое внешней камеры чашки.
При анализе берется 2 — 3 параллельных пробы.
Герметически закрытые чашки оставляют стоять при комнатной температуре на 18 — 20 часов. Далее избыток серной кислоты во внутренней камере чашки оттитровывают 0,01-н раствором едкого натра до перехода окраски из фиолетовой в зеленую.
Параллельно проводят опыт с реактивами в тех же условиях.
Расчет:
| X = | (а — б) x К x 0,14 x 25 x 1000 x 2,143 | , |
| 1 x 50 |
где X — количество мочевины, в мг/л вытяжки; а — количество 0,01-н раствора едкого натра, израсходованное на титрование контроля, мл; б — количество 0,01-н раствора едкого натра, израсходованное на титрование испытуемого раствора, мл; К — поправочный коэффициент для приведения концентрации раствора едкого натра к точно 0,01-н; 0,14 — коэффициент пересчета 0,01-н серной кислоты на азот, мг; 2,143 — коэффициент пересчета азота на мочевину, мг.
ОПРЕДЕЛЕНИЕ САЛИЦИЛОВОЙ КИСЛОТЫ
Реакция основана на том, что салициловая кислота образует с хлорным железом фиолетовое окрашивание. Окрашивание, по-видимому, обусловлено образованием внутрикомплексных солей.
Реактивы
1. Железо хлорное, 1% раствор (свежеприготовленный).
2. Кислота серная, 20% раствор.
3. Натрий углекислый, насыщенный раствор.
4. Спирт этиловый, 40%.
5. Эфир петролейный.
6. Эфир серный.
Ход определения
50 — 100 мл вытяжки нейтрализуют раствором углекислого натрия, после чего подкисляют 20% раствором серной кислоты, вводя избыток ее приблизительно в 1 мл. При необходимости жидкость фильтруют. Переводят раствор в делительную воронку, добавляют 25 — 50 мл смеси равных количеств (объемов) серного и петролейного эфиров и проводят извлечение путем осторожного неоднократного перевертывания воронки (40 — 50 раз) во избежание образования стойкой эмульсии. Дают жидкостям разделиться, затем нижний водный слой спускают через кран воронки и выбрасывают; верхний — эфирный слой — выливают через верхний край воронки в фарфоровую чашку и фильтруют через сухой складчатый фильтр в другую фарфоровую чашку, избегая попадания на фильтр возможно оставшихся капелек воды в эфире. Эфирную вытяжку выпаривают досуха при комнатной температуре в вытяжном шкафу (огнеопасно!). Остаток после выпаривания эфира обрабатывают 10 — 20 каплями разведенного спирта (около 40%), тщательно споласкивая стенки чашки, после чего прибавляют 1 — 2 капли 1% свежеприготовленного хлорного железа. В присутствии салициловой кислоты получается фиолетовое окрашивание.
ОПРЕДЕЛЕНИЕ СТИРОЛА
Метод основан на нитровании стирола с последующим колориметрическим определением окрашенных в желтый цвет соединений в присутствии аммиака. Метод позволяет обнаружить стирол в количестве 0,015 мг в объеме, взятом для определения (0,075 мг/л).
Реактивы
1. Нитроционная смесь: растворяют 10 г азотнокислого аммония в 10 мл серной кислоты (уд. в. 1,835 — 1,84).
2. Аммиак, 25% раствор.
3. Нейтрализованная нитрационная смесь. 1 объем нитрационной смеси (реактив 1) смешивают с 3 объемами дистиллированной воды, нейтрализуют полученный раствор 25% аммиаком до слабощелочной реакции по лакмусу.
4. Четыреххлористый углерод, перегнанный.
5. Стандартный раствор стирола в четыреххлористом углероде. Для его приготовления употребляют свежеперегнанный раствор стирола. 1 мл стандартного раствора равен 50 гамма стирола.
Ход определения
К 200 мл испытуемой вытяжки в делительной воронке прибавляют 10 мл перегнанного четыреххлористого углерода и производят извлечение стирола путем неоднократного перевертывания воронки (50 — 60 раз), повторяют эту операцию троекратно, беря каждый раз по 10 мл растворителя. Соединенные вытяжки четыреххлористого углерода помещают в фарфоровую чашечку и испаряют при комнатной температуре до 2 — 3 капель.
К остатку в чашечке добавляют 1 мл нитрационной смеси и осторожно вращательными движениями перемешивают в течение 5 — 7 минут.
Затем содержимое чашечки количественно переносят в колориметрическую пробирку с притертой пробкой с помощью 3 мл дистиллированной воды.
Кислый раствор в пробирке нейтрализуют 25% аммиаком до слабощелочной реакции по лакмусу, прибавляя аммиак по каплям из бюретки. Нейтрализацию проводят при охлаждении раствора (работу проводят в вытяжном шкафу). В присутствии стирола отмечается наличие ясного желтого окрашивания.
Для количественного определения стирола в фарфоровые чашечки вносят определенное количество стандартного раствора стирола (табл. 9а) и по 1 мл нитрационной смеси. Затем аналогично проводят определение с испытуемым раствором.
Таблица 9а
СТАНДАРТНАЯ ШКАЛА ДЛЯ ОПРЕДЕЛЕНИЯ СТИРОЛА
| Реактив | Номер пробирки | ||||||
| 0 | 1 | 2 | 3 | 4 | 5 | 6 | |
| Стандартный раствор, мл | 0 | 0,3 | 0,4 | 0,5 | 0,6 | 0,7 | 0,8 |
| Содержание стирола, в гамма | 0 | 15 | 20 | 25 | 30 | 35 | 40 |
Примечание. Учитывая высокое токсическое действие четыреххлористого углерода и стирола на организм человека, все работы с ними следует производить в вытяжном шкафу.
Уравнивания растворов производят нейтрализованной нитрационной смесью. Параллельно следует ставить контрольный опыт с реактивами при тех же условиях.
Определение стирола колориметрическим методом
Метод основан на экстракции стирола из водных растворов хлороформом с последующим колориметрическим определением окрашенного в коричневый цвет продукта взаимодействия мономера с формалинсерным реактивом.
Чувствительность метода 0,05 мг/л.
Определению мешает присутствие других ароматических соединений.
Реактивы и аппаратура
Стандартный раствор стирола в хлороформе 0,1 мг/мл.
Формалинсерный реактив. Смешивают 1 мл 37% раствора формальдегида, 100 мл серной кислоты (плотность 1,84).
Хлороформ, х.ч. Хлороформ не должен окрашивать формалинсерный реактив при встряхивании в делительной воронке; в противном случае его нужно очистить перегонкой или промывкой несколькими порциями серной кислоты до прекращения окрашивания.
Воронки делительные емкостью 50 и 250 мл.
Колбы Бунзена.
Фильтры-воронки со стеклянной пористой пластинкой.
Водоструйный насос.
Фотоэлектроколориметр ФЭК-Н-57.
Построение градуировочных графиков
Для построения градуировочных кривых в мерные колбы на 100 мл вносят различные количества стандартного раствора стирола так, чтобы содержание его составляло 0,005; 0,01; 0,02; 0,05 мг в зависимости от предполагаемых количеств мономера в вытяжках. Доводят растворы до 100 мл модельной средой, тщательно перемешивают и далее обрабатывают растворы так же, как вытяжки (см. ход определения). Затем измеряют оптическую плотность окрашенных растворов при лямбда = 453 ммк (синий светофильтр) и для каждой имитирующей среды строят графики зависимости D = f(C), где С — концентрация стирола (в мг/л).
Ход определения
В делительной воронке емкостью 250 мл встряхивают 100 мл вытяжки с 25 мл хлороформа в течение 1 минуты и дают смеси расслоиться. Нижний (хлороформный) слой сливают в делительную воронку емкостью 50 мл и вносят в нее пипеткой 5 мл формалинсерного реактива. Интенсивно взбалтывают содержимое воронки в течение 1 минуты, дают постоять 5 минут для разделения слоев и отделяют окрашенный слой кислоты, сливая его в фильтр-воронку N 3, вставленный при помощи резиновой пробки в колбу Бунзена, которая присоединена к водоструйному насосу. Фильтрование проводят под небольшим вакуумом, окрашенный раствор собирают в пробирку (раствор N 1).
Одновременно проводят холостой опыт со всеми реактивами, но с использованием вместо вытяжки равного объема чистой имитирующей среды. Сразу же после фильтрования определяют оптическую плотность полученного окрашенного раствора N 1 по отношению к раствору холостого опыта, пользуясь для этого кюветами с расстоянием между стенками 1 см. Работа проводится на фотоэлектроколориметре при лямбда = 453 ммк (синий светофильтр). Концентрацию стирола находят по соответствующему градуировочному графику <*> зависимости D = f(C).
<*> При высоких концентрациях стирола (оптическая плотность выше 0,54) можно брать для анализа 25 — 50 мл вытяжки, разбавляя ее до 100 мл соответствующей модельной средой.
Определение стирола при исследовании изделий из полистирола блочного марки «Т» и суспензионного марки «ПС-С» спектрофотометрическим методом
Метод основан на измерении оптической плотности гексанового раствора стирола в ультрафиолетовой области спектра при длине волны лямбда макс = 247 ммк с последующим количественным определением стирола по градуировочному графику.
Мешают определению другие вещества, поглощающие свет так же, как и стирол в ультрафиолетовой области спектра при длине волны 247 ммк.
Чувствительность метода 4 гамма во взятом для исследования объеме вытяжки, ошибка метода 8%.
Реактивы и аппаратура
1. Спектрофотометр СФ-4А или другой марки.
2. Стирол, х.ч., перегнанный перед употреблением. В колбу объемом около 100 мл с отводной трубкой (типа Вюрца) и пришлифованной пробкой помещают 5 — 10 мл стирола, закрывают притертой пробкой, ставят в глицериновую баню и соединяют с маленьким холодильником Либиха, конец которого опускают в небольшую склянку, имеющую притертую пробку. В глицериновую баню помещают термометр на 200 — 250 град. и нагревают ее до температуры 170 град. C. При этой температуре бани отгоняют 2 — 3 мл стирола, который и используют для приготовления стандартного раствора стирола.
3. Спирт этиловый 96 град., перегнанный.
4. Стандартный раствор стирола: в мерную колбу емкостью 25 — 50 мл наливают 10 — 15 мл этилового спирта, закрывают пробкой и взвешивают на аналитических весах. Затем в колбу вносят 2 — 3 капли перегнанного перед употреблением стирола и вновь взвешивают. По разности в весе определяют количество стирола и высчитывают содержание его в 1 мл раствора. Объем раствора в колбе доводят до метки этиловым спиртом. Из приготовленного раствора (N 1) <*> в день построения калибровочного графика готовят рабочий стандартный раствор. Для этого в другую колбу емкостью 100 мл вливают 15 — 20 мл спирта и вносят такое количество раствора N 1, которое соответствует 1 мг стирола. Объем раствора в колбе доводят до метки этиловым спиртом (раствор N 2). 1 мл раствора N 2 содержит 0,01 мг стирола (или 10 гамма).
<*> Раствор N 1 следует хранить в холодильнике.
Построение калибровочного графика
Для построения калибровочного графика в ряд делительных воронок объемом 250 мл вносят по 50 мл бидистиллированной воды и добавляют следующие количества спиртового рабочего раствора стирола N 2 (1 мл = 10 гамма): 0,4; 0,6; 0,8; 1,0; 1,4; 1,8; 2,0 мл; затем стенки каждой воронки смывают 50 мл бидистиллированной воды и извлекают стирол н-гексаном, как указано ниже при определении стирола в вытяжке из исследуемого изделия.
Одновременно в 7 делительных воронок вносят по 100 мл бидистиллированной воды и добавляют спирт в количестве, соответствующем содержанию его во взятом стандартном растворе, а именно: 0,4; 0,6; 0,8; 1,0; 1,4; 1,8; 2,0 мл.
Далее содержимое каждой воронки обрабатывают н-гексаном, как указано ниже. Полученные гексановые экстракты служат эталоном при определении оптической плотности в гексановых вытяжках, содержащих известные количества стирола.
Оптическую плотность измеряют при длине волны 247 ммк в кювете с толщиной слоя 1 см. Полученная оптическая плотность соответствует количеству стирола в 1 мл гексановой вытяжки (например, 0,4 гамма для раствора из первой делительной воронки).
По полученным данным строят калибровочный график, откладывая на оси абсцисс концентрацию стирола в гамма/мл, а на оси ординат — оптическую плотность.
Ход определения
100 — 200 мл вытяжки из исследуемого изделия помещают в делительную воронку, добавляют точно 6 мл н-гексана и извлекают стирол путем осторожного взбалтывания в течение 3 минут, затем еще добавляют точно 4 мл н-гексана и снова взбалтывают в течение 3 минут. После разделения жидкостей (около 30 минут) сливают нижний водный слой как ненужный, а верхний слой — гексановую вытяжку — осторожно сливают через верхний край воронки в сухую пробирку с притертой пробкой (раствор N 3) <*>.
<*> Во избежание попадания в вытяжку воды гексановую вытяжку следует сливать не до конца.
В другую воронку вносят 100 — 200 мл контрольного модельного раствора, примененного при получении вытяжки из изделия, и обрабатывают его 10 мл н-гексана аналогично исследуемой пробе. Гексановый экстракт сливают в пробирку с притертой пробкой (раствор N 4). Каждый из растворов (N 3 и N 4) переносят в кювету с толщиной слоя 1 см и измеряют оптическую плотность раствора N 3 на спектрофотометре при длине волны 247 ммк, при этом кювета с раствором N 4 служит в качестве эталона.
Количество стирола, отвечающее найденной оптической плотности, находят по калибровочному графику.
Содержание стирола (X) в исследуемой вытяжке (в мг/л) высчитывают по формуле:
| X = | C x 10 x 1000 | = | C x 10 | , |
| V x 1000 | V |
где C — количество стирола в гамма/мл гексанового экстракта, найденное по калибровочному графику; V — объем вытяжки, взятой для определения стирола, мл; 10 — количество гексана, взятого для извлечения стирола из исследуемой вытяжки, мл.
Принимая во внимание, что целый ряд других органических веществ может поглощать свет в ультрафиолетовой области спектра при длине волны 247 ммк, в случае обнаружения оптической плотности в гексановой вытяжке из исследуемого раствора при длине волны 247 ммк рекомендуется провести проверку идентичности спектральной характеристики для вещества, обнаруженного в исследуемой вытяжке, со спектральной характеристикой стирола, полученной на чистом растворе стирола.
Для этого в делительную воронку наливают 100 (200) мл модельного раствора (не контактировавшего с исследуемым изделием), 3 мл стандартного раствора стирола (1 мл = 10 гамма) и проводят экстракцию н-гексаном, как указано выше. Гексановую вытяжку сливают в сухую пробирку с притертой пробкой (раствор N 5).
В растворах N 3 и N 5 определяют оптическую плотность в интервале длин волн 230 — 270 ммк.
В качестве эталона используют раствор N 4.
Полученные данные выражают графически в виде кривых светопоглощения, откладывая на оси абсцисс длины волны в ммк, на оси ординат — оптические плотности. Кривые поглощения являются спектральной характеристикой данного вещества.
Растворы стирола в н-гексане имеют максимум поглощения в интервале длин волн 243 — 248 ммк.
Идентичность полученных кривых позволяет сделать окончательное заключение о наличии стирола.
Определение стирола в спиртовой вытяжке из изделий из сополимера СНП-2П, пластифицированного дибутилсебацинатом, спектрофотометрическим методом
Метод основан на определении оптической плотности спиртовой вытяжки при лямбда = 246 ммк.
Чувствительность метода 0,025 мг/л.
Реактивы и аппаратура
1. Стандартный раствор стирола в 40% этиловом спирте, 0,1 мг/мл.
2. Спектрофотометр.
3. Кюветы цилиндрические длиной 50 мм.
Построение градуировочного графика
На спектрофотометре при лямбда = 246 ммк определяют оптическую плотность стандартных растворов стирола в 40% этиловом спирте с концентрацией 0,025; 0,05; 0,10 мг/л и т.д., после чего строят график зависимости D = f(C), где С — концентрация стирола (в мг/л).
Содержание стирола в 40% спиртовых вытяжках находят по оптической плотности, пользуясь градуировочным графиком.
Определение стирола в масляной вытяжке из полистирольных изделий спектрофотометрическим методом
Метод основан на извлечении стирола из масляной вытяжки методом азеотропной разгонки и последующем определении оптической плотности азеотропного дистиллята на спектрофотометре СФ-4 при лямбда = 248 ммк.
Чувствительность метода 0,05 мг/л.
Реактивы и аппаратура
Стандартный раствор стирола в масле, 2,5 мг/л.
Диэтилдитиокарбамат натрия (ДЭДТКН), 0,01% раствор в метиловом спирте (применяется как ингибитор).
Метиловый спирт, дважды перегнанный.
Перегонная установка со шлифами (рис. 6).
Колбы Эрленмейера емкостью 250 и 50 мл с пришлифованными пробками.
Цилиндры мерные емкостью 50 мл с пришлифованными пробками.
Воронки стеклянные.
Спектрофотометр СФ-4.
Построение градуировочного графика
Для построения градуировочного графика готовят серию стандартных растворов стирола в подсолнечном масле с содержанием 0,025; 0,05; 0,1; 0,2; 0,3; 0,6; 0,8; 1,0 мг/л. Затем все растворы при встряхивании последовательно смешивают с 50 мл раствора ДЭДТКН в метиловом спирте и 50 мл дистиллированной воды и подвергают отгонке (см. ход определения). Измеряют оптическую плотность полученных дистиллятов и строят график D = f(C) (рис. 7, кривая 1).
Отрезок m на оси ординат соответствует оптической плотности контрольной пробы относительно дважды перегнанного метилового спирта. Контрольное определение необходимо проводить при каждом анализе, так как различные партии подсолнечного масла и метилового спирта могут сильно отличаться по оптической плотности. Для работы более удобен график зависимости ДЕЛЬТА D = f(C) (рис. 7, кривая 2), на котором по оси ординат откладывают разность оптических плотностей пробы и контроля.
Ход определения
В колбу Эрлейнмейера емкостью 250 мл помещают 50 мл масляной вытяжки, приливают 50 мл раствора ДЭДТКН в метиловом спирте и энергично встряхивают колбу с содержимым в течение 1 минуты. Затем добавляют 50 мл дистиллированной воды и вновь встряхивают. Смесь не должна расслаиваться, в связи с чем она готовится непосредственно перед отгонкой. Приготовленную таким образом смесь заливают в колбу перегонной установки, опустив в нее 30 — 40 нитевидных капилляров длиной 6 — 7 см для равномерного кипения. Перегонную колбу быстро, во избежание расслаивания смеси, помещают в горячую водяную баню с начальной температурой 75 град. C, соединяют с холодильником и отгоняют 50 мл азеотропной смеси стирола с метиловым спиртом. Перегонка начинается при 74,5 = 75 град. C, в конце процесса перегонки температура в колбе не должна превышать 87,5 — 88 град. C. Дистиллят собирают в мерный цилиндр, а затем переносят в колбу емкостью 50 мл с пришлифованной пробкой.
Оптическую плотность дистиллята определяют на спектрофотометре СФ-4 в кварцевых кюветах длиной 50 мм (при лямбда = 248 ммк). Оптическую плотность контрольной и исследуемой проб замеряют относительно дважды перегнанного метилового спирта.
Концентрацию стирола в исследуемой масляной вытяжке находят по градуировочному графику ДЕЛЬТА D = f(С) (рис. 7, кривая 2), предварительно вычислив разницу между оптической плотностью исследуемой и контрольной проб (Dn — Dk). Контрольной пробой служит дистиллят, отогнанный из смеси равных объемов (50 мл) чистого масла, 1% раствора ДЭДТКН в метиловом спирте и воды.
Примечание. Спектрофотометрический метод чрезвычайно чувствителен к чистоте перегонной установки. В случае выброса масла из перегонной колбы в холодильник всю систему следует тщательно отмыть от следов масла и пропарить. Перед анализом для чистки системы необходимо перегнать 40 — 50 мл метилового спирта.
ОПРЕДЕЛЕНИЕ ТРИЭТИЛЕНТЕТРАМИНА С ЭОЗИНОМ К В ВОДНЫХ ВЫТЯЖКАХ
Чувствительность данного метода 0,005 мг триэтилентетрамина в колориметрируемом объеме, или 1 мг/л.
Реактивы
1. Раствор эозина К: 0,02 г эозина К растворяют в 50 мл дистиллированной воды, затем 20 мл этого раствора разбавляют водой до 50 мл.
2. Раствор сернокислой меди: 0,02 г сернокислой меди растворяют в 200 мл воды.
3. Реакционная смесь: 40 мл разбавленного раствора эозина К смешивают с 60 мл раствора сернокислой меди.
4. Стандартный водный раствор триэтилентетрамина, содержащий 0,1 мг в 1 мл.
Ход определения
В колориметрическую пробирку наливают 5 мл водной вытяжки из исследуемого изделия, вносят 5 мл реакционной смеси и взбалтывают. При наличии триэтилентетрамина появляется розовая окраска, которую сравнивают со стандартной шкалой, приготовленной в аналогичных условиях.
При количествах триэтилентетрамина более 0,20 — 0,25 мг окраска неустойчива. В подобных случаях определение проводят после соответствующего разбавления исследуемой вытяжки.
Таблица 10
СТАНДАРТНАЯ ШКАЛА ДЛЯ ОПРЕДЕЛЕНИЯ ТРИЭТИЛЕНТЕТРАМИНА В ВОДНЫХ ВЫТЯЖКАХ
| Реактив | Номер пробирки | |||||||
| 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | |
| Стандартный раствор триэтилентетрамина (1 мл = 0,1 мг), мл | 0 | 0,05 | 0,10 | 0,20 | 0,40 | 0,60 | 0,80 | 1,00 |
| Вода дистиллированная, мл | 5,0 | 4,95 | 4,9 | 4,8 | 4,6 | 4,4 | 4,2 | 4,0 |
| Триэтилентетрамин, мг | 0 | 0,005 | 0,01 | 0,02 | 0,04 | 0,06 | 0,08 | 0,1 |
ОПРЕДЕЛЕНИЕ УРОТРОПИНА
Реакция основана на разложении уротропина в кислой среде до формальдегида с последующим его определением. Из одной молекулы уротропина образуется 6 молекул формальдегида.
Чувствительность 0,25 мг/л.
Реактивы
Кислота фосфорная, 25% раствор.
Ход определения
Водную вытяжку в количестве 100 мл подкисляют 20 мл 25% фосфорной кислоты и перегоняют с водяным паром, как это описано при определении формальдегида. Отгоняют 150 мл дистиллята и проводят в нем определение формальдегида.
ОПРЕДЕЛЕНИЕ ФЕНОЛА
Качественное определение фенола
Реакция основана на взаимодействии водного раствора фенола с бромом с образованием кристаллического осадка трибромфенола в форме игл или пучков из игл.
При наличии незначительного количества фенола и при избытке бромной воды выпадает трибромфенол в смеси с бромистым трибромфенолом (под микроскопом иглы и чешуйчатые пластинки).
Чувствительность — уже при концентрации 1:50000 при длительном стоянии выделяется микрокристаллический осадок.
Реактивы
1. Бромная вода, насыщенный раствор.
2. Винная или щавелевая кислота.
3. Эфир серный.
4. Натрий углекислый, насыщенный раствор.
Ход определения
100 мл вытяжки помещают в круглодонную колбу прибора емкостью 500 мл, снабженную пришлифованной пробкой с двумя Г-образными трубками, конец одной из которых доходит почти до дна колбы, другой заканчивается почти под пробкой (конец ее в колбе имеет длину 3 — 5 см). Первая трубка служит для соединения с парообразователем встык, чтобы пар не соприкасался с каучуком (это должно соблюдаться при всех соединениях стеклянных трубок с помощью каучука). Вторая трубка соединяет колбу с шариковым холодильником, поставленным вертикально. Соединение это осуществляется также при помощи пришлифованной к холодильнику пробки, имеющейся на конце трубки. Нижний конец холодильника опускается в приемник — небольшую эрленмейеровскую колбу, содержащую несколько миллилитров дистиллированной воды, соединенную при помощи пробки и Г-образной стеклянной трубки с другой такой же колбой.
Парообразователем может служить большая стеклянная колба емкостью 1,5 — 2 л, приспособленная соответствующим образом для получения пара и снабженная стеклянной трубкой для уравнивания давления.
Когда все части прибора для перегонки с водяным паром подготовлены и соединены (кроме парообразователя), а парообразователь нагрет, колба с объектом исследования помещена в холодную водяную баню, содержимое колбы быстро подкисляют виннокаменной или щавелевой кислотой, после чего колбу быстро соединяют с парообразователем и начинают нагревать водяную баню под колбой с объектом исследования и парообразователь. Перегонка должна проводиться по возможности медленно. Отгоняют 100 мл дистиллята.
Полученный дистиллят подщелачивают раствором углекислого натрия, переводят в делительную воронку и производят извлечение эфиром. Для этого к раствору прибавляют 10 — 15 мл эфира, воронку перевертывают 40 — 50 раз, избегая сильного взбалтывания, чтобы не образовалась стойкая эмульсия. Извлечение производят 3 раза. Соединенные эфирные вытяжки фильтруют через сухой складчатый фильтр и испаряют в фарфоровой чашечке при комнатной температуре в вытяжном шкафу (огнеопасно!). Остаток после испарения эфира растворяют в 2 — 3 каплях воды и проводят с ними микрореакцию с бромной водой. Каплю раствора из чашечки переносят на предметное стекло, а затем к ней добавляют каплю бромной воды. При наличии фенола выпадает желтовато-белый осадок; при незначительных количествах фенола осадок выпадает при стоянии через некоторое время (приблизительно 10 — 15 минут). При рассмотрении под микроскопом видны мелкие иглы, часто группирующиеся в пучки и звездчатые скопления, возможно выпадение вместе с иглами и чешуйчатых пластинок.
При микроскопическом исследовании сравнивают полученные кристаллы с препаратом, приготовленным из разведенного раствора фенола.
Если необходимо, определяют фенол количественно.
Количественное определение фенола по Архангелову
Метод основан на образовании нитрофенолов при действии азотной кислоты на водные растворы фенола.
При действии на фенол очень разбавленной азотной кислоты получается смесь орто- и паранитрофенолов. Менее разбавленная кислота дает динитрофенол и тринитрофенолпикриновую кислоту — C6H2(NO2)3OH.
Чувствительность 1 мг/л.
Реактивы
1. Азотная кислота, концентрированная.
2. Кали едкое, 50% раствор.
Ход определения
В стаканчик помещают 10 мл дистиллята, прибавляют 10 капель концентрированной азотной кислоты и жидкость доводят до кипения. По охлаждении подщелачивают до щелочной реакции раствором КОН, беря приблизительно на 10 мл жидкости 1 мл 50% раствора КОН. При наличии фенола раствор окрашивается в желтый цвет.
Жидкость из стаканчика переливают в центрифужную пробирку, в которой и производят определение по табл. 11.
Таблица 11
| Окрашивание при рассматривании сбоку | Окрашивание при рассматривании сверху вниз | Окрашивание при рассматривании сверху вниз под углом 45 град. | Содержание фенола, в мг, в 10 мл жидкости |
| Нет | Нет | Едва заметное | 0,01 |
| -«- | Едва заметное | Очень слабо выраженное | 0,02 |
| -«- | Очень слабо выраженное | Слабо выраженное | 0,03 |
| -«- | Слабо выраженное | Ясно выраженное | 0,04 |
| Едва заметное | Ясно выраженное, бледно-желтое | Желтоватое | 0,05 |
| Слабо выраженное | Бледно-желтое | Слабо-желтое | 0,06 |
| Бледно-желтое | Желтоватое | Желтое | 0,07 |
Определение фенола по реакции с 4-аминоантипирином
Метод позволяет определять суммарное содержание фенолов в вытяжках. Фенолы определяются по реакции с 2-аминоантипирином в щелочной среде в присутствии феррацианида калия, являющегося окислителем. При этом образуется соединение типа индофенола, окрашенное в интенсивный красный цвет. Чувствительность метода 0,5 мкг в 5 мл вытяжки, или 0,1 мг/л.
Реактивы
4-аминоантипирин, 2% водный раствор.
Феррицианид калия, 8% водный раствор.
Буферный раствор: 20 г хлорида аммония растворяют в 100 мл концентрированного раствора аммиака, pH этого раствора 9,8.
Натр едкий, 10% водный раствор.
Серная кислота, разбавленный раствор (1:3 по объему).
Стандартный раствор фенола. В мерную колбу емкостью 50 мл наливают 10 — 15 мл дистиллированной воды, взвешивают на аналитических весах, затем помещают кристаллик свежеперегнанного фенола, взвешивают вторично и доводят объем водой до метки. Рассчитывают содержание фенола в 1 мл раствора. Из полученного основного раствора соответствующим разбавлением готовят непосредственно перед его использованием раствор, содержащий 5 мкг/мл (5 мг/л) фенола.
Ход определения
20 мл вытяжки нейтрализуют по индикаторным бумажкам растворами едкого натра или серной кислоты и доводят дистиллированной водой до объема 25 мл в мерной колбе. Из этого раствора отбирают пипеткой 5 мл и переносят в пробирку. Одновременно готовят шкалу стандартов (табл. 12). В пробирки шкалы и исследуемой вытяжки добавляют по 0,5 мл буферного раствора и по 0,2 мл растворов феррицианида калия и 4-аминоантипирина; тщательно перемешивают после добавления каждого реактива и сравнивают полученную окраску со стандартной шкалой или определяют оптическую плотность на фотоколориметре с синим или зеленым светофильтром (лямбда макс = 500 ммк) в кювете с толщиной слоя 10 мм. Эталоном служит раствор, полученный в холостом опыте с 5 мл дистиллированной воды, к которой прибавляют все реактивы, введенные в исследуемую вытяжку.
Таблица 12
СТАНДАРТНАЯ ШКАЛА ДЛЯ ОПРЕДЕЛЕНИЯ ФЕНОЛОВ
| Реактивы | Номер стандартов | |||||||
| 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | |
| Стандартный раствор фенола, мл | 0 | 0,25 | 0,5 | 0,75 | 1,0 | 1,25 | 1,5 | 1,75 |
| Дистиллированная вода, мл | 5,0 | 4,75 | 4,5 | 4,25 | 4,0 | 3,75 | 3,5 | 3,25 |
| Буферный раствор | Во все пробирки по 0,5 мл | |||||||
| Раствор феррицианида калия | Во все пробирки по 0,2 мл | |||||||
| Раствор 4-аминоантипирина | Во все пробирки по 0,2 мл | |||||||
| Содержание фенолов, в мкг, в пробе | 0 | 1,25 | 2,5 | 3,75 | 5,0 | 6,25 | 7,5 | 8,75 |
Для количественного определения зависимости оптических плотностей растворов шкалы стандартов (табл. 12) от концентрации фенола в растворе строят градуировочный график: на оси абсцисс откладывают содержание фенола в мкг, на оси ординат — оптическую плотность.
Содержание фенола в мг/л (X) вычисляют по формуле:
| X = | а x 25 x 1000 | = а x 0,25, |
| 5 x 20 x 1000 |
где а — количество фенолов, найденное по калибровочному графику или по стандартной шкале в 5 мл пробы, в мкг.
Определение фенола по реакции с диазотированным п-нитроанилином
Метод позволяет определить суммарное содержание фенолов в вытяжках. При взаимодействии фенолов с диазотированным п-нитроанилином в щелочной среде появляется окраска от желто-зеленого цвета при низких концентрациях до красно-коричневого при высоких концентрациях. Чувствительность метода 0,5 мкг в 5 мл вытяжки, или 0,1 мг/л.
Реактивы
Натрий углекислый, 4% водный раствор.
Соляная кислота (уд. вес 1,16).
П-нитроанилин, чистый.
Нитрит натрия, свежеприготовленный 2% водный раствор.
Натр едкий, 10% водный раствор.
Серная кислота, разбавленная (1:3 по объему).
Ход определения
20 мл вытяжки нейтрализуют по индикаторным бумажкам растворами едкого натра или серной кислоты и доводят дистиллированной водой до объема 25 мл в мерной колбе. Из этого раствора отбирают пипеткой 5 мл и переносят в пробирку. Одновременно готовят шкалу стандартов (табл. 13).
Таблица 13
СТАНДАРТНАЯ ШКАЛА ДЛЯ ОПРЕДЕЛЕНИЯ ФЕНОЛА
| Реактивы | Номер стандарта | ||||||||
| 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | |
| Стандартный раствор фенола, мл | 0 | 0,25 | 0,5 | 0,75 | 1,0 | 1,25 | 1,5 | 1,75 | 2,0 |
| Дистиллированная вода, мл | 5,0 | 4,75 | 4,5 | 4,25 | 4,0 | 3,75 | 3,5 | 3,25 | 3,0 |
| Раствор углекислого натрия | Во все пробирки по 1,0 мл | ||||||||
| Диазотированный п-нитроанилин | Во все пробирки по 0,2 мл | ||||||||
| Содержание фенола, в мкг, в пробе | 0 | 1,25 | 2,5 | 3,75 | 5,0 | 6,25 | 7,5 | 8,75 | 10 |
В пробирки шкалы и исследуемой вытяжки добавляют по 1 мл раствора углекислого натрия и по 0,2 мл раствора диазотированного п-нитроанилина. Тщательно перемешивают и через 5 минут сравнивают интенсивность полученной окраски со стандартной шкалой или определяют оптическую плотность на фотоколориметре с синим светофильтром (лямбда макс = 470 ммк) в кювете с толщиной слоя 10 мм. Эталоном служит раствор, полученный в холостом опыте с 5 мл дистиллированной воды, к которой прибавляют все реактивы, введенные в исследуемую вытяжку.
Для количественного определения зависимости оптической плотности растворов шкалы стандартов (табл. 13) от концентрации фенола строят градуировочный график: на оси абсцисс откладывают содержание фенола в мкг, на оси ординат — оптическую плотность.
Содержание фенола в мг/л (X) вычисляют по формуле:
| X = | а x 25 x 1000 | = а x 0,25, |
| 5 x 20 x 1000 |
где а — количество фенолов, найденное по калибровочному графику или по стандартной шкале в 5 мл пробы, в мкг.
ОПРЕДЕЛЕНИЕ ФОРМАЛЬДЕГИДА
Определение формальдегида с фуксиносернистой кислотой (реакция Шиффа)
Метод основан на дистилляции формальдегида из вытяжки с помощью водяного пара и на его реакции в сильнокислой среде с фуксиносернистой кислотой с образованием сине-фиолетового окрашивания.
Чувствительность метода 0,002 мг в колориметрируемом объеме, или 0,6 мг/л.
Метод специфичен, другие альдегиды в количестве до 0,3 мг (300 гамма) не мешают определению.
Определение формальдегида проводят немедленно после получения вытяжки, так как при хранении ее формальдегид может улетучиться.
Реактивы
1. Стандартный раствор формальдегида. Предварительно из официнального раствора формалина (формалин — водный раствор формальдегида) готовят более слабый раствор, количество формальдегида в котором определяют йодометрически.
5 мл формалина продажного препарата помещают в мерную колбу на 250 мл, доводят содержимое колбы до метки водой и хорошо перемешивают (раствор N 1). 5 мл раствора N 1 переносят в коническую колбу с притертой пробкой емкостью 200 — 250 мл, добавляют из бюретки 40 мл 0,1-н раствора йода и тотчас добавляют по каплям 30% раствор едкого натра до образования устойчивого бледно-желтого окрашивания. Колбу помещают в темное место и оставляют на 10 минут. Затем содержимое колбы подкисляют 5 мл 10% соляной или серной кислоты и снова ставят раствор на 10 минут в темное место. По истечении этого времени в раствор вливают 150 мл дистиллированной воды и титруют 0,1-н раствором гипосульфита до слабо-желтого цвета раствора. Далее добавляют 1 мл 0,5% раствора крахмала и продолжают титрование до исчезновения синей окраски. Одновременно проводят холостой опыт с теми же реактивами и в тех же условиях, при этом вместо 5 мл раствора N 1 берут 5 мл дистиллированной воды. Разница между количеством гипосульфита (мл), израсходованного при титровании в холостом опыте и при титровании испытуемого раствора, соответствует количеству йода (мл), израсходованного на окисление формальдегида.
Количество формальдегида (X) в 1 мл разбавленного раствора формалина (раствор N 1) высчитывают по следующей формуле:
| X = | (a — b) x K x 1,50 | , |
| 5 |
где X — количество формальдегида в 1 мл разбавленного раствора формалина (раствор N 1), мг; b — количество гипосульфита, израсходованного на титрование исследуемого раствора, мл; a — количество гипосульфита, израсходованного на титрование в холостом опыте, мл; K — поправочный коэффициент для приведения концентрации раствора гипосульфита к точно 0,1-н; 1,50 — количество формальдегида, эквивалентное 1 мл точно 0,1-н раствора гипосульфита, мг.
Далее, исходя из полученных данных, готовят раствор формальдегида с содержанием 0,1 мг в 1 мл.
Пример. Содержание формальдегида в приготовленном растворе формалина (раствор N 1) равно 8,04 мг в 1 мл. Для приготовления 500 мл раствора с содержанием 0,1 мг в 1 мл (раствор N 2) следует взять 50/8,04 = 6,22 мл оттитрованного раствора N 1.
Раствор сохраняется в течение 1,5 месяца.
Из полученного раствора (N 2), содержащего 0,1 мг формальдегида в 1 мл, готовят соответствующим разбавлением рабочие стандартные растворы, содержащие 0,01 и 0,001 мг формальдегида в 1 мл. Растворы должны быть свежеприготовленными.
2. Йод, 0,1-н раствор.
3. Гипосульфит натрия (Na2S2O3 x 5H2O), 0,1-н раствор.
4. Фуксиносернистая кислота (реактив Шиффа). 0,025% водный раствор основного фуксина готовят из растертого в порошок кристаллического препарата путем растворения взятой навески в горячей воде. Раствор фильтруют теплым. После охлаждения в раствор пропускают слабый ток сернистого ангидрида до слабо-розовой окраски раствора, после чего раствор оставляют до следующего дня. Если обесцвечивания раствора не наступает в течение суток, добавляют к раствору по каплям водный раствор сернистого ангидрида до обесцвечивания или до слабого розового оттенка (избыток сернистого ангидрида неблагоприятно сказывается на чувствительности реакции).
Реактив хранят в темной склянке с хорошо притертой пробкой. Сернистый газ получают следующим способом: в колбу с отводной трубкой наливают концентрированную серную кислоту, закрывают пробкой с капельной воронкой, содержащей 40% раствор кислого сернистокислого натрия. При добавлении раствора кислого сернистокислого натрия по каплям к серной кислоте получают непрерывный ток сернистого газа.
Для получения водного раствора сернистой кислоты ток сернистого газа пропускают через две склянки, из которых первая содержит 300 мл прокипяченной дистиллированной воды, а вторая — 200 мл. Склянки охлаждают проточной водой или водой со льдом.
Реактив должен быть бесцветным или слегка желтоватым.
5. Натр едкий, 30% водный раствор.
6. Крахмал, 0,5% раствор (свежеприготовленный и отфильтрованный).
7. Соляная кислота, концентрированная (уд. в. 1,19).
8. Соляная кислота 10%.
Ход определения <*>
<*> В водной и уксуснокислых вытяжках формальдегид определяют без отгона.
100 мл испытуемой вытяжки помещают в отгоночную круглодонную колбу аппарата для перегонки с водяным паром емкостью около 500 мл и отгоняют 150 мл дистиллята. Колба с дистиллятом при перегонке должна быть погружена в воду со льдом.
5 мл дистиллята переносят в колориметрическую пробирку. Одновременно готовят шкалу стандартов (табл. 14).
Таблица 14
СТАНДАРТНАЯ ШКАЛА ДЛЯ ОПРЕДЕЛЕНИЯ ФОРМАЛЬДЕГИДА С ФУКСИНОСЕРНИСТОЙ КИСЛОТОЙ
| Реактив | Номер пробирки | |||||
| 0 | 1 | 2 | 3 | 4 | 5 | |
| Стандартный раствор (1 мл = 0,01 мг), мл | 0 | 0,20 | 0,40 | 0,60 | 0,80 | 1,00 |
| Дистиллированная вода, мл | 5,0 | 4,8 | 4,6 | 4,4 | 4,2 | 4,0 |
| Содержание формальдегида, мг | 0 | 0,002 | 0,004 | 0,006 | 0,008 | 0,01 |
В пробирки шкалы и исследуемого раствора приливают по 0,5 мл концентрированной соляной (уд. вес 1,19) и по 1 мл фуксиносернистой кислоты, содержимое перемешивают и оставляют на 40 — 50 минут.
При наличии формальдегида образуется интенсивное синее или сине-фиолетовое окрашивание. Интенсивность полученной окраски в испытуемой пробирке сравнивают со стандартной шкалой.
Параллельно необходимо ставить контрольный опыт с реактивами при тех же условиях.
Определение формальдегида с солянокислым фенилгидразином по Шриверу
Реакция основана на том, что при прибавлении фенилгидразина к раствору, содержащему формальдегид, в присутствии окислителя (K3Fe(CH)6) происходит окисление фенилгидразина до фенилгидразона, причем промежуточный продукт этого окисления конденсируется с формальдегидом, образуя вещество, окрашенное в оранжево-красный цвет.
Определение формальдегида проводят немедленно после получения вытяжки, так как при хранении ее формальдегид может улетучиться.
Реактивы
1. Фенилгидразин солянокислый (C6H5 x NH2 x HCl), 1% водный раствор (свежеприготовленный и профильтрованный).
2. Калий железосинеродистый (K3Fe(CH)6), 5% водный раствор.
3. Соляная кислота, концентрированная.
Ход определения
Перегонку формальдегида осуществляют из вытяжки с водяным паром и в полученном дистилляте проводят его определение.
К 10 мл дистиллята прибавляют 2 мл раствора солянокислого фенилгидразина и перемешивают. Затем приливают 1 мл железосинеродистого калия, перемешивают, добавляют 5 мл концентрированной соляной кислоты и снова перемешивают. В присутствии формальдегида появляется оранжево-красное окрашивание.
При незначительных количествах формальдегида (1 — 10 мкг в 10 мл) раствор имеет окраску от желтовато-розовой до интенсивно-розовой.
Чувствительность 0,1 мг/л.
При испытании уксуснокислой вытяжки рекомендуется проводить холостой опыт с модельным раствором при равных условиях проведения реакции с вытяжкой для исключения возможного наличия уксусного альдегида в уксусной кислоте. Уксусный альдегид дает слабую окраску при концентрации 1:500 — 1:1000.
Реакция Шривера более чувствительна по сравнению с реакцией Шиффа (с фуксиносернистой кислотой).
При слабо положительной реакции Шривера количество формальдегида может быть определено приближенно по табл. 15.
Таблица 15
ПРИБЛИЖЕННОЕ ОПРЕДЕЛЕНИЕ ФОРМАЛЬДЕГИДА С ПОМОЩЬЮ СОЛЯНОКИСЛОГО ФЕНИЛГИДРАЗИНА
| Окрашивание при рассматривании сбоку | Окрашивание при рассматривании сверху вниз | Окрашивание при рассматривании сверху вниз под углом 45 град. | Содержание формальдегида в мг/л |
| Слабо-розоватожелтая опалесценция | Розовато-желтая опалесценция | Очень слабо выраженная розоватожелтая окраска | 0,1 |
| Слабо выраженная желтоваторозовая окраска | Розовая с желтоватым оттенком окраска | Слабо-розовое | 0,3 |
| Розовое | Розовое | Интенсивно-розовое | 0,5 |
| Розовое | Интенсивно-розовое | Розовое с красноватым оттенком | 0,5 |
| Интенсивно-розовое | Розовое с красноватым оттенком | Розовато-красное | 0,9 |
Определение формальдегида в вытяжках <*> по реакции с хромотроповой кислотой
<*> В водной и уксусной вытяжках формальдегид определяют без отгона.
Метод основан на дистилляции формальдегида из вытяжек с помощью водяного пара и его цветной реакции с хромотроповой кислотой.
Метод позволяет обнаружить содержание формальдегида в количестве 0,2 гамма в колориметрируемом объеме, или 0,1 мг/л.
Другие альдегиды мешают определению при количествах порядка десятых долей мг. Определение формальдегида проводят немедленно после получения вытяжки, так как при хранении ее формальдегид может улетучиться.
Реактивы
1. Серная кислота, концентрированная (уд. вес 1,835 — 1,84).
2. Хромотроповая кислота (1,8-диоксинафталин-3,6-дисульфокислота) или ее динатриевая соль (C10H6O8S2Na2), 1% водный раствор, свежеприготовленный.
3. Стандартный раствор формальдегида. Приготовление см. выше.
Ход определения
100 мл испытуемой вытяжки помещают в круглодонную колбу аппарата для перегонки с водяным паром и далее проводят дистилляцию, как указано выше.
3,0 мл дистиллята переносят в колориметрическую пробирку с притертой пробкой, приливают 0,4 мл 1% водного раствора хромотроповой кислоты, 1,7 мл концентрированной серной кислоты (уд. в. 1,835 — 1,84) и перемешивают. Пробирку помещают в кипящую водяную баню на 30 минут. При охлаждении пробирки перемешивают ее содержимое и наблюдают окрашивание через 40 — 50 минут.
В зависимости от содержания формальдегида появляется более или менее интенсивное красно-фиолетовое окрашивание, которое сравнивают со стандартной шкалой (табл. 16), приготовленной одновременно в аналогичных условиях, или измеряют оптическую плотность раствора на фотоэлектроколориметре при длине волны 540 ммк в кювете с толщиной слоя 10 мм. Эталоном служит раствор от холостого опыта. Количество формальдегида, отвечающее найденной оптической плотности, находят по градуировочному графику, который готовят, пользуясь той же стандартной шкалой (табл. 16).
Таблица 16
СТАНДАРТНАЯ ШКАЛА ДЛЯ ОПРЕДЕЛЕНИЯ ФОРМАЛЬДЕГИДА С ХРОМОТРОПОВОЙ КИСЛОТОЙ
| Реактив | Номер пробирки | |||||
| 0 | 1 | 2 | 3 | 4 | 5 | |
| Стандартный раствор формальдегида (1 мл = 0,001 мг = 1 гамма), мл | 0 | 0,20 | 0,40 | 0,60 | 0,80 | 1,00 |
| Дистиллированная вода, мл | 3,0 | 2,8 | 2,6 | 2,4 | 2,2 | 2,00 |
| Содержание формальдегида, в гамма | 0 | 0,2 | 0,4 | 0,6 | 0,8 | 1,00 |
Определение формальдегида в водной и уксусной вытяжках по реакции с хромотроповой кислотой
1. Стандартный раствор формальдегида. Приготовление см. выше.
2. Хромотроповая кислота. Приготовление см. выше.
Ход определения
2 мл уксуснокислой или водной вытяжки переносят в стеклянную пробирку с притертой пробкой. В 6 таких же пробирках готовят стандартную шкалу, как указано в табл. 17.
Таблица 17
СТАНДАРТНАЯ ШКАЛА ДЛЯ ОПРЕДЕЛЕНИЯ ФОРМАЛЬДЕГИДА
| Реактив | Номер пробирки | |||||
| 0 | 1 | 2 | 3 | 4 | 5 | |
| Стандартный раствор формальдегида (1 мл = 0,001 мг = 1 гамма), мл | 0 | 0,2 | 0,4 | 0,6 | 0,8 | 1,0 |
| Уксусная кислота, 1% раствор, мл | 2,0 | 1,8 | 1,6 | 1,4 | 1,2 | 1,0 |
| Содержание формальдегида, в гамма | 0 | 0,2 | 0,4 | 0,6 | 0,8 | 1,0 |
Во все пробирки шкалы и пробы добавляют по 0,4 мл 1% раствора хромотроповой кислоты или ее динатриевой соли, перемешивают и добавляют 1,7 мл концентрированной серной кислоты. Пробирки закрывают пробками, перемешивают содержимое пробирок и помещают их в кипящую водяную баню на 30 минут. Затем вынимают пробирки из бани и оставляют их при комнатной температуре на 40 — 60 минут.
В зависимости от количества формальдегида растворы окрашиваются в более или менее интенсивный красно-фиолетовый цвет.
Колориметрирование проводят, сравнивая на белом фоне окраску испытуемого раствора с окраской стандартных растворов, или проводят определение с помощью фотоэлектроколориметра при длине волны 540 ммк в кювете с толщиной слоя 10 мм.
Для построения градуировочного графика измеряют оптические плотности растворов той же стандартной шкалы (табл. 17 или 18).
Таблица 18
СТАНДАРТНАЯ ШКАЛА ДЛЯ ОПРЕДЕЛЕНИЯ ФОРМАЛЬДЕГИДА
| Реактив | Номер пробирки | ||||||
| 0 | 1 | 2 | 3 | 4 | 5 | 6 | |
| Стандартный раствор формальдегида (1 мл = 0,01 мг = 10 гамма), мл | 0 | 0,05 | 0,10 | 0,20 | 0,30 | 0,40 | 0,50 |
| Уксусная кислота, 1% раствор, мл | 2,00 | 1,95 | 1,90 | 1,80 | 1,70 | 1,60 | 1,50 |
| Содержание формальдегида, в гамма | 0 | 0,5 | 1,0 | 2,0 | 3,0 | 4,0 | 5,0 |
При высоком содержании формальдегида готовят вторую шкалу, как указано в табл. 18.
Метод позволяет обнаружить содержание формальдегида в количестве 0,2 гамма в колориметрируемом объеме.
ОПРЕДЕЛЕНИЕ ФТАЛЕВОЙ КИСЛОТЫ И ФТАЛЕВОГО АНГИДРИДА
Определение основано на получении фенолфталеина путем конденсации (в присутствии серной кислоты) фталевого ангидрида с фенолом. Возникновение малиново-красной окраски в щелочной среде свидетельствует о наличии фталевой кислоты или фталевого ангидрида.
Реактивы
1. Кислота серная, концентрированная.
2. Натр или кали едкое, 50% раствор.
3. Спирт этиловый.
4. Фенол. К 100 г расплавленного на водяной бане фенола (кристаллической карболовой кислоты) приливают при помешивании 10 мл дистиллированной воды.
5. Глицерин.
Ход определения
100 мл водной или NaCl-вытяжки из исследуемых изделий выпаривают на водяной бане в фарфоровом тигле досуха. К сухому остатку в тигле добавляют одну каплю реактива N 4, 1 — 2 капли концентрированной серной кислоты и далее проводят определение, как указано в методе определения дибутилфталата и диоктилфталата.
Чувствительность метода 2 мг/л.
ОПРЕДЕЛЕНИЕ ЭМУЛЬГАТОРА ОП-10 В ВОДНОЙ ВЫТЯЖКЕ
Метод основан на гидролизе ОП-10 концентрированной серной кислотой с последующим колориметрическим определением окрашенного производного октилфенола с м-нитробензальдегидом. Определению мешают высшие спирты и альдегиды.
ОП-10 (C8H17C6-H4-O-(CH2CH2O)C10H2CH2(OH)) — продукт конденсации окиси этилена с октилфенолом.
Чувствительность метода 2 гамма в колориметрируемом объеме (0,2 мг/л).
Реактивы
1. Серная кислота (уд. вес 1,82 — 1,84).
2. М-нитробензальдегид. Для анализа применяют свежеприготовленный раствор: 0,1 г м-нитробензальдегида растворяют в 10 мл концентрированной серной кислоты.
3. Приготовление стандартного раствора ОП-10. Отвешивают в маленьком стаканчике (с точностью до 0,1 мг) 10 — 15 мг ОП-10; приливают 5 мл концентрированной серной кислоты и стеклянной палочкой тщательно перемешивают содержимое стаканчика. После того как все будет тщательно перемешано, навеску ОП-10 в серной кислоте переносят через воронку в мерную колбу емкостью 25 мл, смывая стаканчик 10 — 15 мл концентрированной серной кислоты. Осторожным вращательным движением колбы производят перемешивание ее содержимого в течение 20 минут при 20 — 25 град. C.
За это время происходит омыление ОП-10.
По окончании омыления раствор в колбе доводят до метки концентрированной серной кислотой.
Рассчитывают содержание ОП-10 в мг/мл.
Путем соответствующего разведения готовят стандартные растворы с содержанием 0,1 мг в 1 мл и 0,01 мг в 1 мл ОП-10.
Ход определения
25 мл водной вытяжки выпаривают досуха на водяной бане в фарфоровой чашечке. К сухому остатку приливают 2 мл концентрированной серной кислоты, обмывая всю поверхность чашечки. 1 мл раствора переносят в колориметрическую пробирку. Одновременно готовят стандартную шкалу, как указано в табл. 19.
Таблица 19
СТАНДАРТНАЯ ШКАЛА ДЛЯ ОПРЕДЕЛЕНИЯ ОП-10
| Реактив | Номер пробирки | |||||||||
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | |
| Стандартный раствор ОП-10 (0,1 мг ОП-10 в мл), мл | — | — | — | — | — | 0,2 | 0,4 | 0,6 | 0,8 | 1,0 |
| Стандартный раствор ОП-10 (0,01 мг ОП-10 в мл), мл | 0,2 | 0,4 | 0,6 | 0,8 | 1,0 | — | — | — | — | — |
| Серная кислота, мл | 0,8 | 0,6 | 0,4 | 0,2 | 0 | 0,8 | 0,6 | 0,4 | 0,2 | 0 |
| Содержание ОП-10, гамма | 2 | 4 | 6 | 8 | 10 | 20 | 40 | 60 | 80 | 100 |
Во все пробирки шкалы и испытуемых проб наливают по 0,2 мл 1% раствора м-нитробензальдегида. Содержимое в пробирках встряхивают и помещают на пять минут в водяную баню при температуре 90 — 100 град. C. Затем сравнивают окраску проб со шкалой на белом фоне.
ОПРЕДЕЛЕНИЕ ЭПИХЛОРГИДРИНА
Определение эпихлоргидрина с фуксиносернистой кислотой
Метод основан на дистилляции эпихлоргидрина из вытяжек с помощью водяного пара и на способности его окисляться йодной кислотой с образованием формальдегида, определяемого с фуксиносернистым реактивом.
Чувствительность метода 0,01 мг эпихлоргидрина в колориметрируемом объеме.
Определению мешают формальдегид, окись этилена, этиленгликоль, этиленхлоргидрин.
Реактивы
1. Периодат калия (KJO4) или натрия (NaJO4), 3% раствор в 5% растворе серной кислоты. Периодат калия растворяется при нагревании до кипения.
2. Серная кислота, 1:3.
3. Сульфит натрия (Na2SO3 x 7H2O), 10% раствор, свежеприготовленный.
4. Фуксиносернистая кислота (реактив Шиффа).
5. Стандартный раствор эпихлоргидрина. Готовят основной раствор: в мерную колбу на 50 мл наливают 20 — 25 мл дистиллированной воды и взвешивают на аналитических весах. Вносят в колбу с водой 2 — 3 капли эпихлоргидрина и вновь взвешивают. По разности веса определяют количество эпихлоргидрина. Раствор в колбе доводят до метки дистиллированной водой. Высчитывают содержание эпихлоргидрина в 1 мл раствора.
Путем соответствующего разведения готовят перед употреблением рабочий стандартный раствор с содержанием эпихлоргидрина 0,1 мг и 0,02 мг в 1 мл.
Ход определения
100 мл исследуемой вытяжки помещают в круглодонную колбу аппарата для перегонки с водяным паром емкостью около 300 мл и отгоняют 50 мл дистиллята. Колба с дистиллятом при перегонке должна быть погружена в воду со льдом.
К 5,0 мл дистиллята в колориметрической пробирке с притертой пробкой приливают 1 мл серной кислоты (1:3) и 0,25 мл 3% раствора периодата калия. Пробирку закрывают пробкой, содержимое перемешивают и ставят в кипящую водяную баню на 5 минут для окисления эпихлоргидрина. По охлаждении содержимого пробирки избыток йодной кислоты разрушают 10% сульфитом натрия, добавляя его по каплям до обесцвечивания выделившегося йода, затем прибавляют 1 мл фуксиносернистого реактива, содержимое пробирки перемешивают и через 40 минут — 1 час измеряют интенсивность сине-фиолетового окрашивания, пользуясь стандартной шкалой (табл. 20), приготовленной одновременно с пробой в аналогичных условиях. Параллельно необходимо ставить контрольный опыт с реактивами при тех же условиях.
Таблица 20
СТАНДАРТНАЯ ШКАЛА ДЛЯ ОПРЕДЕЛЕНИЯ ЭПИХЛОРГИДРИНА
| Реактив | Номер пробирки | |||||
| 0 | 1 | 2 | 3 | 4 | 5 | |
| Стандартный раствор эпихлоргидрина (1 мл = 0,1 мг), мл | 0 | 0,10 | 0,20 | 0,30 | 0,40 | 0,50 |
| Дистиллированная вода, мл | 5,0 | 4,90 | 4,80 | 4,70 | 4,60 | 4,50 |
| Содержание эпихлоргидрина, мг | 0 | 0,01 | 0,02 | 0,03 | 0,04 | 0,05 |
Определение эпихлоргидрина с хромотроповой кислотой в водной, солевой (NaCl) и виннокислой вытяжках
Метод основан на дистилляции эпихлоргидрина из вытяжек с помощью водяного пара и на его окислении йодной кислотой до формальдегида с последующим проведением цветной реакции с хромотроповой кислотой.
Метод позволяет обнаружить содержание эпихлоргидрина в количестве 0,001 мг в колориметрируемом объеме.
Определению мешают формальдегид, этиленгликоль, окись этилена.
Реактивы
1. Серная кислота: 10% раствор (по весу), раствор 1:1 (по объему).
2. Периодат калия (KJO4), 1,5% раствор в серной кислоте (уд. в. 1,83 — 1,84). Растворение проводят при нагревании.
3. Сульфит натрия (Na2SO3 x 7H2O), 10% водный раствор, свежеприготовленный.
4. Хромотроповая кислота или ее динатриевая соль. 0,15 г реактива растворяют в 5 мл 10% раствора серной кислоты, затем прибавляют 125 мл серной кислоты (уд. в. 1,83 — 1,84), если нужно, фильтруют через стеклянный фильтр. Свежеприготовленный раствор.
5. Стандартный раствор эпихлоргидрина. Приготовление см. выше.
Ход определения
100 мл испытуемой вытяжки помещают в круглодонную колбу аппарата для перегонки с водяным паром и отгоняют 50 мл дистиллята. Колба с дистиллятом при перегонке должна быть погружена в воду со льдом. 2 мл дистиллята (из мерной колбы на 50 мл) переносят в колориметрическую пробирку, добавляют 0,5 мл раствора серной кислоты (1:1) и 0,3 мл раствора периодата калия, перемешивают и оставляют на 30 минут при комнатной температуре.
Для восстановления избытка периодата калия в пробирку добавляют по каплям раствор сульфита натрия до обесцвечивания выделяющегося йода, затем приливают 2,5 мл раствора хромотроповой кислоты, осторожно перемешивают. Пробирку помещают в кипящую водяную баню на 30 минут. К охлажденному раствору приливают 3 мл дистиллированной воды и перемешивают. При разбавлении раствора водой коричневая окраска исчезает и остается фиолетовая окраска, характерная для продукта реакции формальдегида с хромотроповой кислотой. Через 5 минут интенсивность окраски исследуемого раствора сравнивают со стандартной шкалой (табл. 21), приготовленной одновременно с пробой в аналогичных условиях. Параллельно необходимо ставить контрольный опыт с реактивами при тех же условиях.
Таблица 21
СТАНДАРТНАЯ ШКАЛА ДЛЯ ОПРЕДЕЛЕНИЯ ЭПИХЛОРГИДРИНА
| Реактив | Номер пробирки | |||||||
| 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | |
| Стандартный раствор эпихлоргидрина (0,02 мг/мл), мл | 0 | 0,05 | 0,1 | 0,2 | 0,3 | 0,4 | 0,6 | 0,8 |
| Дистиллированная вода, мл | 2 | 1,95 | 1,9 | 1,8 | 1,7 | 1,6 | 1,4 | 1,2 |
| Содержание эпихлоргидрина, мг | 0 | 0,001 | 0,002 | 0,004 | 0,006 | 0,008 | 0,012 | 0,016 |
ОПРЕДЕЛЕНИЕ ЭТИЛЕНГЛИКОЛЯ
Метод основан на перегонке этиленгликоля из вытяжек с бензолом и на последующем определении в дистилляте этиленгликоля путем окисления его периодатом калия или натрия в сернокислом растворе до формальдегида. Формальдегид определяется колориметрическим методом с помощью фуксиносернистой или хромотроповой кислоты.
Метод специфичен в присутствии метилового и этилового спирта.
Дистилляция этиленгликоля из вытяжек
В круглодонную колбу аппарата (рис.  емкостью 100 мл помещают 20 мл исследуемой вытяжки и приливают 50 мл бензола. Присоединяют колбу к вертикально поставленному холодильнику с помощью специального приемника-ловушки, служащего для улавливания воды вместе с этиленгликолем (ГОСТ 1594-69). Колбу нагревают на водяной бане.
емкостью 100 мл помещают 20 мл исследуемой вытяжки и приливают 50 мл бензола. Присоединяют колбу к вертикально поставленному холодильнику с помощью специального приемника-ловушки, служащего для улавливания воды вместе с этиленгликолем (ГОСТ 1594-69). Колбу нагревают на водяной бане.
При этом бензол, увлекаемая им вода и этиленгликоль конденсируются и попадают в приемник-ловушку, из которого бензол возвращается обратно в колбу.
Перегонка длится 12 — 16 часов; обычно отгоняют 9 — 10 мл дистиллята.
По окончании перегонки прибор разбирают. Дистиллят из ловушки переносят в делительную воронку, отделяют водный слой от бензола и определяют в водном слое этиленгликоль с функсиносернистой или хромотроповой кислотами (рис. 9, 10).
Определение этиленгликоля с фуксиносернистой кислотой
Метод определения основан на окислении этиленгликоля периодатом калия или натрия в сернокислом растворе до формальдегида с последующим колориметрическим определением его по реакции с фуксиносернистой кислотой.
Метод позволяет обнаружить содержание этиленгликоля в количестве 0,008 мг, или 8 гамма, в колориметрируемом объеме, или 0,8 мг/л.
Реактивы
1. Периодат калия (KJO4) или натрия (NaJO4), 3% раствор в 5% растворе серной кислоты. Периодат натрия растворяют в серной кислоте на холоду, периодат калия — при нагревании до кипения.
2. Серная кислота, 5% раствор (по объему).
3. Сульфит натрия (Na2SO3 x 7H2O), 10% раствор, свежеприготовленный.
4. Фуксиносернистый реактив. Приготовление см. выше.
5. Стандартный раствор этиленгликоля. Готовят исходный раствор: в мерную колбу емкостью 50 мл наливают 20 — 25 мл дважды перегнанной дистиллированной воды и взвешивают на аналитических весах. Вносят в колбу с водой 1 — 2 капли перегнанного этиленгликоля (температура кипения 195 град.) и вновь взвешивают. По разности веса определяют количество этиленгликоля. Раствор в колбе доводят до метки водой. Высчитывают содержание этиленгликоля в 1 мл раствора.
Путем соответствующего разведения готовят перед употреблением рабочий стандартный раствор с содержанием этиленгликоля 0,02 мг и 0,01 мг в 1 мл.
Стандартный раствор, содержащий несколько мг этиленгликоля в 1 мл, сохраняется без изменения в течение 6 месяцев.
Ход определения
5 мл дистиллята вносят в колориметрическую пробирку с притертой пробкой. Одновременно готовят шкалу стандартов (табл. 22).
Таблица 22
СТАНДАРТНАЯ ШКАЛА ДЛЯ ОПРЕДЕЛЕНИЯ ЭТИЛЕНГЛИКОЛЯ
| Реактив | Номер пробирки | ||||
| 0 | 1 | 2 | 3 | 4 | |
| Стандартный раствор этиленгликоля, мл | 0 | 0,40 | 0,60 | 0,80 | 1,00 |
| Дистиллированная вода, мл | 5,00 | 4,60 | 4,40 | 4,20 | 4,00 |
| Содержание этиленгликоля, мг | 0 | 0,008 | 0,012 | 0,016 | 0,02 |
В пробирки шкалы и исследуемого раствора приливают по 1 мл 5% раствора серной кислоты и по 0,25 мл 3% раствора периодата калия или натрия. Содержимое пробирок встряхивают и оставляют на 30 минут. Затем избыток йодной кислоты разрушают 10% раствором сульфита натрия, добавляя его по каплям до обесцвечивания выделившегося йода. Далее добавляют по 1 мл фуксиносернистой кислоты. Содержимое пробирок перемешивают и оставляют стоять на 30 — 40 минут.
В присутствии этиленгликоля в зависимости от его количества появляется более или менее интенсивная фиолетовая или голубая окраска. Интенсивность окраски исследуемого раствора сравнивают со шкалой.
Параллельно необходимо ставить контрольный опыт с реактивами при тех же условиях.
Определение этиленгликоля с хромотроповой кислотой
Метод основан на окислении этиленгликоля периодатом калия или натрия в сернокислом растворе до формальдегида с последующим определением его по реакции с хромотроповой кислотой.
Чувствительность метода 0,001 мг (или 1 гамма) в колориметрируемом объеме.
Реактивы
1. Серная кислота (уд. в. 1,84), разбавленная 1:1 и 10% раствор.
2. Периодат калия (KJO4), 1,5% раствор в серной кислоте (уд. в. 1,84), растворяют при нагревании.
3. Сульфит натрия (Na2SO3 x 7H2O). 50 г растворяют при нагревании в 50 мл воды. Пригоден к употреблению в течение трех суток.
4. Хромотроповая кислота или ее динатриевая соль.
5. Стандартный раствор этиленгликоля. Приготовление см. выше.
Ход определения
2 мл дистиллята переносят в колориметрическую пробирку. Одновременно готовят шкалу стандартов (табл. 23). В пробирки шкалы и в пробирку с дистиллятом добавляют по 0,5 мл серной кислоты (1:1) и по 0,3 мл раствора периодата калия, перемешивают и оставляют на 30 минут при комнатной температуре. Для восстановления избытка периодата калия в пробирки добавляют по каплям раствор сульфита натрия. После добавления первой капли сульфита натрия содержимое пробирки хорошо встряхивают, при этом начинает выделяться йод. При дальнейшем добавлении сульфита натрия выделение йода возрастает (окраска раствора делается более интенсивной). После полного восстановления периодата калия добавляемый сульфит натрия реагирует с выделившимся йодом. Сульфит натрия добавляют осторожно по каплям до исчезновения окраски раствора от выделившегося йода (избегать избытка, сульфит натрия мешает реакции), затем приливают по 2,5 мл раствора хромотроповой кислоты, осторожно перемешивают и помещают пробирки в кипящую водяную баню на 30 минут. Вынимают пробирки из бани и после охлаждения раствора добавляют по 3 мл воды, перемешивают и наблюдают окрашивание растворов через 15 — 20 минут. В присутствии этиленгликоля в зависимости от его количества появляется более или менее интенсивная красно-фиолетовая или розовая окраска раствора.
Таблица 23
СТАНДАРТНАЯ ШКАЛА ДЛЯ ОПРЕДЕЛЕНИЯ ЭТИЛЕНГЛИКОЛЯ
| Реактив | Номер пробирки | |||||||
| 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | |
| Стандартный раствор этиленгликоля (1 мл = 0,01 мг), мл | 0 | 0,1 | 0,2 | 0,3 | 0,4 | 0,6 | 0,8 | 1,0 |
| Содержание этиленгликоля, мг | 0 | 0,001 | 0,002 | 0,003 | 0,004 | 0,006 | 0,008 | 0,01 |
Интенсивность окраски в пробирке с дистиллятом сравнивают со шкалой.
ОПРЕДЕЛЕНИЕ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ, ВХОДЯЩИХ В РЕЦЕПТУРУ ИССЛЕДУЕМОГО ИЗДЕЛИЯ
ОПРЕДЕЛЕНИЕ ЖЕЛЕЗА
Реакция основана на взаимодействии в сильнокислой среде окисного железа и роданида аммония с образованием окрашенного в красный цвет роданида железа — Fe(CNS)3.
Реактивы
1. Аммиак, 10% раствор.
2. Аммоний роданистый, 50% раствор.
3. Кислота соляная, концентрированная.
4. Кислота соляная, 10% раствор.
5. Пергидроль.
Ход определения
К 10 мл вытяжки, доведенной до нейтральной реакции по лакмусу, с помощью разведенной соляной кислоты или раствора аммиака прибавляют при взбалтывании 5 капель концентрированной соляной кислоты, 2 — 3 капли пергидроля, затем приливают 0,2 — 0,5 мл 50% роданистого аммония и перемешивают. Появление красного окрашивания указывает на наличие солей железа.
Необходимо проверить реактивы на отсутствие в них железа.
ОПРЕДЕЛЕНИЕ КАЛЬЦИЯ
Определение основано на осаждении щавелевокислого кальция и идентификации последнего с помощью одной из характерных реакций на кальций — осаждения кристаллов гипса (CaSO4 x 2H2O).
Открываемый минимум — 1 гамма кальция.
Реактивы
1. Аммиак, 10% раствор.
2. Аммоний щавелевокислый, насыщенный раствор.
3. Кислоты: соляная, концентрированная; соляная, 10% раствор; соляная, 1% раствор; серная, 1 — 2-н; уксусная, 20% раствор; уксусная, 10% раствор.
4. Метилрот: 0,1 г метилрота растворяют в 60 мл этилового спирта и добавляют дистиллированную воду до 100 мл.
Ход определения
50 мл вытяжки выпаривают в фарфоровой чашке на водяной бане досуха и сухой остаток осторожно озоляют. Золу смачивают несколькими каплями бидистиллята, добавляют 1 — 2 мл концентрированной соляной кислоты и выпаривают досуха. Обработку осадка соляной кислотой повторяют 2 — 3 раза.
Сухой остаток растворяют в 1 — 2 мл 1% соляной кислоты и переносят в центрифужную пробирку, чашку промывают 2 — 3 мл бидистиллята и присоединяют к раствору в пробирке. Если содержимое пробирки мутное, муть отделяют центрифугированием. Прозрачный раствор (3 — 5 мл) переносят в чистую пробирку, добавляют каплю метилрота и по каплям 10% раствор аммиака до слабо-желтого окрашивания. Если при этом образуется муть, ее растворяют путем добавления 20% уксусной кислоты (добавляя по каплям до слабо-розового цвета раствора).
Пробирку с раствором помещают в кипящую водяную баню, через 5 — 10 минут прибавляют 1 — 2 мл горячего раствора щавелевокислого аммония и оставляют пробирку в горячей бане (при слабом подогреве) на 15 — 30 минут. При наличии в исследуемом растворе кальция образуется белый осадок щавелевокислого кальция. Осадок отделяют центрифугированием, растворяют при нагревании в 10% растворе соляной кислоты, затем с полученным раствором проводят микрореакцию следующим способом.
Каплю прозрачного раствора переносят на предметное стекло, осторожно выпаривают на маленьком пламени горелки. Остаток после выпаривания растворяют в капле 10% раствора уксусной кислоты и наносят рядом каплю 10% серной кислоты; дают каплям натечь друг на друга или соединяют их тонкой стеклянной нитью.
В присутствии солей кальция тотчас или через несколько минут, в зависимости от количества кальция в растворе, выпадают кристаллы гипса (CaSO4 x 2H2O) в виде длинных игл, часто группирующихся в пучки, снопики, звездчатые и веерообразные скопления.
При незначительных количествах кальция кристаллизация происходит медленно и главным образом по периферии капли. При более высоких концентрациях через некоторое время возможно выпадение, кроме игл, ромбических пластинок, двойников с входящим углом в виде ласточкина хвоста и X-образных двойников.
ОПРЕДЕЛЕНИЕ КОБАЛЬТА
Роданомеркуриат аммония — (NH4)2Hg(SCN)4 — образует с солями кобальта кристаллическое соединение Co[Hg(SCN)4], окрашенное в синий цвет. Чувствительность реакции 0,2 гамма кобальта.
Реактивы
1. 3 г сулемы и 3,3 г роданистого аммония растворяют в 3 мл H2O. Этот реактив используется также для открытия меди, кадмия и цинка.
2. Уксусная кислота, 1:50.
3. Соляная кислота, 1:1.
4. Соляная кислота, 1% раствор.
Ход определения
50 мл вытяжки выпаривают в фарфоровой чашке на водяной бане досуха, полученный остаток осторожно озоляют. Золу растворяют в соляной кислоте (1:1) и выпаривают досуха.
Остаток после выпаривания растворяют при нагревании в возможно малом количестве 1% раствора соляной кислоты. При наличии нерастворимого осадка его отделяют центрифугированием. Фильтрат выпаривают досуха (удаление HCl), остаток растворяют в нескольких каплях воды и проводят микрореакцию на кобальт с ртутно-родановым реактивом (реактив N 1) на образование роданисто-ртутно-кобальтовой соли.
Для этого каплю раствора из чашки переносят на предметное стекло и осторожно выпаривают на стекле досуха, после чего на полученный остаток после выпаривания наносят еще калю, снова выпаривают и т.д. (наслаивание капель).
Полученный таким образом остаток после выпаривания на стекле растворяют в капле уксусной кислоты (1:50), наносят рядом небольшую каплю ртутно-роданового реактива и дают каплям натечь друг на друга. При наличии соли кобальта выпадают интенсивно-синие кристаллы роданисто-ртутно-кобальтовой соли Co[Hg(SCN)4] ромбической системы. Эти кристаллы по большей части соединяются в неодинаковые группы, образуя шиповидные шарики, розетки, неправильные треугольники, иногда и кустики. Незначительные количества никеля не мешают реакции, но уже в 10-кратном количестве, превышающем кобальт, вредят реакции. При наличии меди одновременно с синими кристаллами роданисто-ртутно-кобальтовой соли выпадают и желто-зеленые кристаллы роданисто-ртутно-медной соли. При значительных количествах цинка и кадмия и при наличии меди выпадают смешанные кристаллы буро-фиолетовой окраски. В присутствии только цинка или кадмия выпадают смешанные кристаллы светло-синего цвета.
ОПРЕДЕЛЕНИЕ КРЕМНЕКИСЛОТЫ
Кремневая кислота с молибдатом аммония образует аммонийную соль кремнемолибденовой кислоты (NH4)4SiMa12O40), которая окисляет бензидин до синего хиноидного продукта при одновременном восстановлении молибдена до «молибденовой сини».
Чувствительность реакции 0,1 гамма SiO2.
Фосфорная и мышьяковая кислоты мешают реакции. Однако аммонийная соль кремнемолибденовой кислоты растворима в азотной кислоте, тогда как аммонийные соли мышьяковомолибденовой и фосфорномолибденовой кислот нерастворимы в ней и могут быть отделены.
Реактивы
1. Молибдат аммония: 5 г молибдата аммония растворяют на холоду в 100 мл воды и раствор приливают к 35 мл азотной кислоты (уд. в. 1,2).
2. Бензидин: 0,5 г бензидина или хлоргидрата бензидина растворяют в 10 мл концентрированной уксусной кислоты и разбавляют водой до 10 мл.
3. Ацетат натрия, насыщенный раствор.
4. Азотная кислота, 6-н раствор.
5. Щавелевая кислота, 1% раствор.
Ход определения
В платиновый тигель помещают 1 каплю исследуемого раствора (кислотность раствора не должна быть выше 0,5-н), добавляют каплю раствора молибденовокислого аммония и нагревают на небольшом пламени горелки до кипения. Далее тигель охлаждают, добавляют 1 каплю раствора бензидина и 1 — 2 капли раствора ацетата натрия. При наличии кремневой кислоты появляется синее окрашивание.
Выполнение реакции в присутствии фосфат-иона: 1 — 2 капли исследуемого раствора помещают в центрифужную пробирку, добавляют 2 — 3 капли раствора молибдата аммония, нагревают на небольшом пламени до кипения, добавляют 2 — 3 капли 6-н раствора азотной кислоты и центрифугируют. Прозрачную жидкость сливают в платиновый тигель и осторожно нагревают на сетке на маленьком пламени горелки до кипения. После охлаждения к содержимому тигля добавляют 2 — 3 капли 1% раствора щавелевой кислоты, 1 — 2 капли раствора бензидина и 2 — 3 капли раствора ацетата натрия. Появление синего окрашивания укажет на присутствие кремневой кислоты.
ОПРЕДЕЛЕНИЕ МАРГАНЦА
Метод основан на окислении марганца персульфатом аммония до марганцевой кислоты в присутствии катализатора AgNO3.
Присутствие хлоридов, бромидов, йодидов и других анионов, связывающих серебро или разрушающих MnO4, мешает реакции.
Чувствительность реакции очень велика. По Ф. Файглю можно открыть 0,1 гамма марганца (в капле раствора 1:500000).
Реактивы
1. Аммоний персульфат [(NH4)2S2O8], 50% раствор, свежеприготовленный.
2. Кислоты: азотная, концентрированная; серная, 20% раствор.
3. Пергидроль.
4. Серебро азотнокислое, 0,1-н раствор.
Ход определения
50 мл вытяжки выпаривают в фарфоровой чашке на водяной бане досуха. Сухой остаток осторожно озоляют (до получения золы белого или серовато-белого цвета). Для ускорения озоления золу можно обработать пергидролем. Золу для удаления хлоридов смачивают концентрированной азотной кислотой и выпаривают на водяной бане досуха. Обработку золы азотной кислотой повторяют. Далее к золе добавляют 5 мл 20% серной кислоты и выпаривают до 1 мл. Обработку серной кислотой повторяют 2 раза. К содержимому фарфоровой чашки добавляют 1 — 2 мл бидистиллята, затем и раствор переносят в центрифужную пробирку. Если раствор мутный, его центрифугируют. К прозрачному раствору, собранному в чистую пробирку, добавляют 1 мл 0,1-н раствора азотнокислого серебра и 1 мл 50% водного раствора персульфата аммония (свежеприготовленного), взбалтывают и помещают в кипящую водяную баню на 15 минут, после чего охлаждают в струе холодной воды. В присутствии марганца появляется розово-фиолетовая окраска (образование марганцевой кислоты).
ОПРЕДЕЛЕНИЕ МЫШЬЯКА
(ГОСТ 5512. Продукты и напитки пищевые и вкусовые. Методы определения мышьяка)
Реактивы
1. Гидразинсульфат.
2. Кислота азотная, концентрированная и 10% раствор.
3. Кислота серная, концентрированная и 1:4 (по объему), не содержащая мышьяка.
4. Мышьяк, стандартный раствор: 13,2 мг перекристаллизованного и очищенного возгонкой мышьяковистого ангидрида растворяют в небольшом количестве 10% раствора едкого натра; раствор переводят в колбу Кьельдаля емкостью 100 мл, смывают остаток из стаканчика небольшим количеством дистиллированной воды в ту же колбу, добавляют 10 мл концентрированной серной кислоты (без мышьяка) и содержимое колбы нагревают, укрепив последнюю на штативе, до появления паров серной кислоты; затем раствор охлаждают и, добавив к нему 0,1 г гидразинсульфата через стеклянную трубку, не доходящую до содержимого колбы на несколько сантиметров, нагревают до кипения, которое поддерживают в течение 10 минут; охлажденный раствор количественно переводят в мерную литровую колбу и добавляют дистиллированную воду до метки; 1 мл такого раствора содержит 0,01 мг, или 10 гамма мышьяка.
5. Олово хлористое в кристаллах.
6. Парафин, 5% раствор в петролейном эфире.
7. Реактив антипириновый: 1 г чистого антипирина и 2 г йодистого калия растворяют в 30 мл дистиллированной воды.
8. Ртуть бромная, 5% спиртовый раствор. Продажную бромную ртуть очищают возгонкой. Для этого ее помещают в фарфоровую чашку и, покрыв воронкой, нагревают на песчаной бане; для охлаждения наружной поверхности воронки ее покрывают слоем мокрой фильтровальной бумаги. Бромная ртуть возгоняется и осаждается на холодных стенках воронки в виде белых игл, 2,5 г очищенной бромной ртути растворяют в 50 мл этилового спирта.
9. Свинец уксуснокислый, 5% раствор.
10. Спирт этиловый.
11. Цинк металлический без мышьяка.
12. Эфир петролейный.
13. Эфир серный.
14. Бромно-ртутные полоски: беззольные фильтры разрезают на полоски необходимых размеров, один конец полоски смачивают спиртовым раствором бромной ртути путем погружения ее в раствор; удаляют избыток раствора с полоски встряхиванием и укладывают на стеклянные палочки для досушивания. Сухие бромно-ртутные бумажки хранят в темной склянке с хорошо притертой пробкой в темном месте.
Все применяемые реактивы и модельные растворы должны быть испытаны на отсутствие в них мышьяка (холостой опыт).
Прибор для выделения мышьяка. Прибор представляет собой колбу А объемом около 50 мл с вертикальной насадкой. Насадка состоит из хорошо пришлифованной к колбе А стеклянной трубки Б длиной 50 — 70 мм, с диаметром просвета 6 — 7 мм и стеклянной трубки В длиною 30 — 40 мм, с диаметром просвета 2 — 2,5 мм, хорошо пришлифованной к верхнему концу трубки Б. Для конструирования прибора можно пользоваться обыкновенной конической колбой объемом около 50 мл, причем отдельные части прибора соединяются с помощью резиновых пробок, в отверстие которых вставляют трубки указанных размеров.
Для поглощения могущего образоваться сероводорода в трубку Б помещают комочек ваты, предварительно смоченный 5% раствором уксуснокислого свинца, отжатый затем между листами фильтровальной бумаги и разрыхленный. Нижний конец трубки Б закрывают рыхлым комочком сухой гигроскопической ваты.
Для фиксации мышьяка в трубку В помещают полоску бромно-ртутной бумажки, наружный конец которой, выступающий свободно из трубки В, загибают для удержания полоски в просвете трубки В. Бромно-ртутные полоски во всех приборах помещают на одинаковую глубину, кроме того, они должны занимать одинаковое положение по отношению к стенкам трубок В.
Одновременно с определением мышьяка готовят эталоны, имея минимум 6 вышеописанных приборов.
Качественное определение мышьяка цинкокислотным методом
Метод основан на восстановлении пятивалентного мышьяка в трехвалентный, а затем в мышьяковистый водород, дающий окрашенное соединение с полоской бромно-ртутной бумаги.
Реакция неспецифична для мышьяка. Сероводород и фосфористый водород дают аналогичную реакцию. Сурьмянистый водород вызывает желтое или серое окрашивание при количествах свыше 0,1 мг на определение.
Ход определения
25 мл исследуемого раствора переносят в колбу А прибора для выделения мышьяка, добавляют равный объем разведенной (1:4) серной кислоты. Далее добавляют 0,2 г хлористого олова в кристаллах, 2 г цинка и тотчас же закрывают колбу насадкой, содержащей бромно-ртутную полоску, помещают колбу в холодную воду и ставят в темное место.
Если в течение часа при хорошей реакции между цинком и серной кислотой (о чем судят по пузырькам газа) бромно-ртутная полоска остается без изменения, реакцию считают отрицательной, а неорганический мышьяк в исследуемой вытяжке — необнаруженным.
При наличии мышьяка бромно-ртутная полоска окрашивается в желтовато-бурый цвет.
При положительной качественной реакции проводят количественное определение мышьяка.
Количественное определение мышьяка
Метод основан на мокрой минерализации органического вещества с помощью серной и азотной кислот с последующим восстановлением пятивалентного мышьяка в трехвалентный, а затем в мышьяковистый водород, дающий окрашенное соединение с полоской бромно-ртутной бумаги. Интенсивность окраски сравнивают с окраской полосок, полученных из растворов с известным количеством мышьяка (от 0,001 до 0,01 мг).
Минерализация органического вещества. 100 мл вытяжки из исследуемого образца переносят в фарфоровую чашку, добавляют едкий натр (10% раствор) до щелочной реакции по лакмусу, выпаривают на кипящей водяной бане до небольшого объема (10 — 15 мл) и переводят с помощью 15 мл 10% азотной кислоты в колбу Кьельдаля объемом около 25 мл. Колбу с исследуемым веществом помещают на асбестовую сетку, укрепляют на штативе и добавляют осторожно 10 мл серной кислоты (концентрированной).
Если исследуемая вытяжка содержит сахаристые вещества, колбу с исследуемым веществом после добавления азотной кислоты помещают в баню с холодной водой, добавляют осторожно при перемешивании, небольшими порциями 10 мл концентрированной серной кислоты и оставляют в холодной воде на 20 — 30 минут. Затем колбу переносят на асбестовую сетку с отверстием, закрытым листочком асбеста.
Выше колбы Кьельдаля на том же штативе укрепляют делительную воронку, наливают в нее концентрированную азотную кислоту, устанавливают носик воронки над центром колбы и нагревают содержимое колбы до кипения. Во время нагревания, при котором вначале выделяются пары воды, а затем красно-бурые окислы азота, пламя горелки регулируют так, чтобы вся колба представлялась окрашенной в красно-бурый цвет выделяющимися окислами азота.
Если по окончании выделения окислов азота жидкость в колбе начнет темнеть, в колбу, не прекращая нагревания, добавляют азотную кислоту из делительной воронки по каплям, отрегулировав кран воронки так, чтобы в минуту вытекало 6 — 8 капель кислоты. Через 15 — 20 минут добавление азотной кислоты прекращают, вынимают из-под колбы асбестовый листочек и содержимое колбы кипятят до появления густых белых паров серной кислоты, после чего кипятят еще 10 минут. Если в течение этого времени жидкость остается бесцветной, то считают минерализацию органического вещества законченной. Если же начинается потемнение жидкости (обугливание органического вещества), то добавляют в колбу азотную кислоту из делительной воронки и продолжают минерализацию, как указано выше.
После полной минерализации органического вещества (узнаваемой по отсутствию потемнения жидкости при 10-минутном кипячении, считая с появления густых белых паров серной кислоты) бесцветную или светло-желтую жидкость охлаждают, разбавляют равным количеством воды и кипятят до появления паров серной кислоты с целью удаления связанных окислов азота.
Восстановление пятивалентного мышьяка в трехвалентный. После удаления связанных окислов азота в колбу Кьельдаля к охлажденной жидкости добавляют 0,2 — 0,4 мл гидразинсульфата, внося его через длинную стеклянную трубку. Это необходимо для того, чтобы гидразинсульфат не остался вне жидкости, на стенках колбы, что может неблагоприятно отразиться на результате анализа. Для полного разложения гидразинсульфата жидкость нагревают до кипения и кипятят в течение 10 минут. По охлаждении содержимое колбы количественно переносят в мерную колбу на 100 мл, ополаскивая колбу дистиллированной водой, охлаждают, добавляют дистиллированную воду до метки и хорошо перемешивают.
Выделение мышьяка. 25 мл исследуемого раствора, полученного после разрушения органического вещества, переносят в колбочку А прибора для выделения мышьяка. В колбочки А приборов N 2, 3, 4, 5 и 6 вносят соответственно 0,1; 0,3; 0,5; 0,7 и 1 мл стандартного раствора мышьяка (1 мл которого равен 0,01 мг Ag) и затем в каждую колбочку добавляют разведенную (1:9) серную кислоту в таком количестве, чтобы общее количество жидкости во всех колбочках было равно 25 мл. Далее в каждую из 6 колбочек вносят 0,2 г хлористого олова в кристалликах, 2 г металлического цинка и тотчас же колбы закрывают насадками, содержащими вату, пропитанную уксуснокислым свинцом, и бромно-ртутную полоску бумаги, после чего приборы помещают в холодную воду.
После растворения цинка (обычно в течение 3,5 — 4 часов) вынимают из трубок В полоски бромно-ртутной бумаги, окрашенные в зависимости от количества мышьяка в цвета от светло-желтого до желто-бурого, отмечают на них номера колбочек, фиксируют окраски, смачивая полоски путем погружения в 5% раствор парафина в петролейном эфире, отжимают между листками фильтровальной бумаги и высушивают на воздухе.
Колориметрирование. Окрашенную полоску из исследуемого раствора сравнивают с окраской полосок, полученных из растворов с известным количеством мышьяка (от 0,001 до 0,01 мг), принимая во внимание окраску обеих поверхностей. Наиболее легко колориметрируются окраски с содержанием мышьяка не более 0,01 мг; большие количества мышьяка затрудняют колориметрирование, и результаты получаются менее точные.
Окрашенные полоски бромно-ртутной бумаги, полученные из известного количества мышьяка, заключают между двумя стеклянными пластинками, края которых заклеивают двойным слоем бумаги. Полученную шкалу окрасок хранят в темном месте и пользуются ею для ориентировочного определения мышьяка в исследуемом растворе.
В надлежащих условиях шкала сохраняется без изменений около 6 месяцев. При изменении окраски полосок шкалу следует приготовить заново.
Для окончательного количественного определения мышьяка проводят выделение его из исследуемого и стандартного растворов одновременно, чтобы сохранить одинаковые условия опыта. Расчет:
| X = | а x 100 x 1000 | , |
| б x в |
где X — содержание мышьяка, в мг/л; а — содержание мышьяка в стандартном растворе, дающем окраску полоски, одинаковую с окраской полоски из испытуемого раствора, в мг; б — количество испытуемого раствора, взятого для выделения мышьяка, в мл; в — количество исследуемой вытяжки, взятое для минерализации, в мл.
ОПРЕДЕЛЕНИЕ СВИНЦА, МЕДИ И ЦИНКА
(ГОСТ 5370-58. Продукты и напитки пищевые и вкусовые. Методы определения)
Отделение свинца, меди и цинка от других элементов и их разделение
Отделение свинца, меди и цинка от других элементов основано на осаждении названных элементов в виде сульфидов и последующего разделения их с помощью характерных для каждого элемента реакций.
Реактивы
1. Аммиак 25%.
2. Аммиак 10%.
3. Железо сернистое.
4. Кислоты: азотная, концентрированная, соляная 10%, соляная 1%, уксусная, концентрированная.
5. Натр едкий 10%.
6. Натрий уксуснокислый, насыщенный раствор, подкисленный уксусной кислотой до слабокислой реакции по лакмусу.
7. Пергидроль.
8. Спирт 96 град.
Подготовка исследуемой вытяжки. 200 мл исследуемой вытяжки выпаривают небольшими порциями в небольшой фарфоровой чашке (диаметром около 7 см) на водяной бане досуха, высушивают на песчаной или воздушной бане, а затем осторожно обугливают и озоляют на слабом огне или в муфеле сначала с открытой дверцей, поместив чашку у самой дверцы муфеля. После обугливания органического вещества чашку переставляют в глубь муфеля и закрывают дверцу. Допускается лишь слабо-красное накаливание стенок муфеля.
После озоления и охлаждения чашки к золе добавляют 5 мл соляной кислоты, разведенной дистиллированной водой 1:1, каплю пергидроля и выпаривают на водяной бане досуха. Сухой остаток обрабатывают 2 мл 10% соляной кислоты, добавляют 3 мл воды и фильтруют через предварительно смоченный фильтр в колбу объемом около 100 мл; фарфоровую чашечку и фильтр промывают 15 мл дистиллированной воды, собирая промывные воды в ту же колбу. В полученный раствор пропускают ток сероводорода, как будет указано ниже.
Выделение и разделение свинца, меди и цинка. Солянокислый раствор нагревают до 40 — 50 град. C и в течение 40 — 60 минут пропускают ток сероводорода. При этом в осадок выпадают сульфиды свинца, олова, меди и т.д., тогда как металлы 1, 2 и 3-й групп остаются в растворе.
Сероводород пропускают через узко оттянутую трубку, доходящую до дна колбы. Выпавший осадок сульфидов и серы отделяют центрифугированием в пробирке емкостью около 10 мл, собирая жидкость в небольшую фарфоровую чашку (раствор А, в котором может быть цинк).
Осадок сульфидов промывают 1 — 2 раза 1% раствором соляной кислоты, насыщенным сероводородом, присоединяя промывную жидкость к раствору А.
К промытому осадку сульфидов тотчас же добавляют 5 капель 10% раствора едкого натра, нагревают в кипящей водяной бане, разбавляют 10 мл воды и центрифугируют (в растворе может быть олово, в осадке — сульфиды свинца и меди, а также сера). При большом осадке сульфидов обработку едким натром производят дважды.
К осадку сульфидов меди и свинца добавляют 5 — 10 капель смеси крепкой серной и азотной кислот, взятых в равных количествах, нагревают на небольшом пламени горелки, осторожно вводя (периодически) дно пробирки в пламя горелки.
Обработку осадка заканчивают после полного удаления паров азотной кислоты и появления белых тяжелых паров серного ангидрида.
После охлаждения в пробирку добавляют 0,5 — 1,0 мл дистиллированной воды и такое же количество спирта. Если после прибавления воды и спирта раствор остается прозрачным, то соли свинца считают необнаруженными. При появлении в растворе мути или выпадении белого осадка (PbSO4) производят следующие операции:
а) осадок сернокислого свинца отделяют центрифугированием, собирая раствор в маленькую фарфоровую чашку.
Осадок промывают 2 — 3 раза небольшим количеством (около 10 мл) разбавленного этилового спирта (1:1 по объему), присоединяя промывные воды к раствору в фарфоровой чашке. В дальнейшем осадок сернокислого свинца обрабатывают, как указано ниже в подпункте «б».
Раствор выпаривают досуха на водяной бане, охлаждают и добавляют 1 — 5 капель 25% раствора аммиака. Появление очень слабого голубого окрашивания указывает на наличие во взятой навеске следов меди (меньше 0,1 мг). При более интенсивном окрашивании добавляют 1 — 2 мл дистиллированной воды и, если раствор оказывается мутным (от гидрата окиси железа), 1 — 5 капель 25% раствора аммиака. Осадок отделяют центрифугированием, сливая раствор в мерный цилиндр объемом 10 мл. Осадок в пробирке промывают 1 — 2 раза небольшим количеством дистиллированной воды, содержащей небольшое количество аммиака (около 1%), присоединяя промывную жидкость к раствору в мерном цилиндре; содержимое цилиндра дистиллированной водой доводят до определенного объема и сохраняют для количественного определения меди (раствор Б);
б) к осадку сернокислого свинца, оставшемуся в центрифужной пробирке, добавляют 1 мл насыщенного раствора уксуснокислого натрия, предварительно слабо подкисленного уксусной кислотой, нагревают в кипящей водяной бане в течение 5 — 10 минут, добавляют 1 мл дистиллированной воды и фильтруют через маленький фильтр, предварительно смоченный дистиллированной водой, собирая фильтрат в мерный цилиндр объемом 10 мл. Пробирку и фильтр промывают несколько раз небольшими порциями дистиллированной воды, собирая промывные воды в тот же цилиндр. К раствору в цилиндре добавляют дистиллированную воду до 10 мл и хорошо перемешивают (раствор В). 5 мл раствора из цилиндра переносят в центрифужную пробирку, добавляют 3 капли двухромовокислого калия (5% раствор) и хорошо перемешивают. Если раствор остается прозрачным в течение 10 минут, считают свинец необнаруженным. При наличии в растворе свинца появляется желтая муть (PbCrO4). В последнем случае проводят количественное определение свинца в растворе, оставшемся в цилиндре (раствор В).
Определение свинца
Для количественного определения свинца 1 мл раствора В из цилиндра переносят в плоскодонную пробирку с делениями на 10 мл; в три другие такие же пробирки вносят стандартный раствор свинца в количестве 0,01; 0,015 и 0,02 мг. В пробирки со стандартным раствором добавляют по 0,1 мл насыщенного раствора уксуснокислого натрия, слабо подкисленного уксусной кислотой. Далее во все 4 пробирки добавляют по 3 капли двухромовокислого калия (5% раствор), хорошо перемешивают и через 10 — 15 минут образовавшуюся муть в пробирке с испытуемым раствором сравнивают со стандартными растворами.
Если для определения свинца берут не 1 мл испытуемого раствора, а меньше или больше, то в пробирки со стандартными растворами свинца добавляют столько уксуснокислого натрия, сколько его содержится во взятом для определения количестве испытуемого раствора. Как в испытуемом, так и в стандартных растворах содержание уксуснокислого натрия должно быть одинаковым. Несоблюдение этих условий приводит к неправильным результатам.
Пример. Мутность испытуемого раствора соответствует мутности стандартного раствора, содержащего 0,01 мг свинца. Для количественного определения свинца было взято 2 мл из 10 мл всего испытуемого раствора, полученного из 200 мл вытяжки. Следовательно, в 1 л содержится:
| X = | 0,01 x 10 x 1000 | = 3,3 мг свинца. |
| 2 x 200 |
Определение меди
Часть или весь раствор Б, судя по качественной реакции, переносят в пробирку для колориметрирования с делениями на 5, 10 и 15 мл (или в обыкновенную пробирку). В три другие пробирки, одинаковые с первой, наливают стандартный раствор меди, содержащий 0,1, 0,3 и 0,5 мг меди. Далее во все 4 пробирки добавляют по 2 мл аммиака (25% раствор) и дистиллированную воду, доводя объем содержимого каждой пробирки до 10 мл, и хорошо перемешивают. Интенсивность окрашивания испытуемого раствора сравнивают с окраской стандартных растворов.
Если окраска испытуемого раствора интенсивнее окраски стандартного раствора, содержащего 0,1 мг меди, и слабее окраски стандартного раствора с содержанием 0,3 мг меди, считают, что в испытуемом растворе количество меди равно 0,2 мг.
Содержание меди (X1) в мг/л вытяжки вычисляют по формуле:
| X1 = | 0,2 x V1 x 1000 | , |
| V x а |
где V — количество исследуемого раствора, взятое для колориметрирования, в мл; V1 — общее количество раствора, исследуемого на медь (раствор Б), в мл; а — количество вытяжки, взятой для определения свинца, меди и цинка, в мл; 0,2 — количество меди, найденное при сравнении испытуемого раствора со стандартным раствором, в мг.
Определение цинка
К раствору А добавляют 5 — 7 капель пергидроля, выпаривают на водяной бане досуха, остаток растворяют в 2 — 3 мл 10% раствора соляной кислоты и нейтрализуют насыщенным раствором соды (Na2CO3) до образования белой мути (выпадают углекислые соли железа, кальция, цинка и других металлов). Для растворения выпавших карбонатов прибавляют (по каплям) 10% раствор соляной кислоты до получения прозрачного раствора.
К прозрачному раствору прибавляют избыток (5 — 7 мл) насыщенного раствора уксуснокислого натрия (CH3COONa), ставят на сетку и нагревают до кипения, чтобы осадить железо. Кипятят в течение 1 — 2 минут, затем горячий раствор немедленно фильтруют через сухой фильтр. Осадок на фильтре промывают 3 раза горячим раствором уксуснокислого натрия (1 мл CH3COONа на 20 мл воды). Фильтрат должен быть прозрачным и бесцветным, в противном случае осаждение железа следует повторить.
В горячий фильтрат в течение 20 минут пропускают ток промытого сероводорода. При наличии цинка выпадает белый хлопьевидный осадок или небольшая белая муть (если цинка мало). Колбу после пропускания сероводорода закрывают пробкой, через 2 — 3 часа осадок отфильтровывают (или центрифугируют) и промывают 2 — 3 раза сероводородной водой.
Промытый осадок сернистого цинка растворяют (в центрифужной пробирке или на маленьком фильтре) 2 мл горячей 40% соляной кислоты. Одну каплю этого раствора переносят на предметное стекло и проводят микрореакцию на цинк.
Каплю солянокислого раствора цинка, помещенную на предметное стекло, осторожно выпаривают досуха на маленьком пламени горелки (не перегревать). К остатку добавляют одну каплю раствора уксусной кислоты (1:50 по объему) и смешивают с сухим остатком. Рядом с этой каплей наносят каплю ртутно-роданистого калия и, соединив их тонкой стеклянной палочкой (нитью), не перемешивая, ставят под микроскоп. При наличии в растворе цинка почти моментально по краям капли выпадает белый осадок в виде перистых крестов и разветвленных групп, напоминающих листья папоротника. В случае положительной микрохимической реакции количественное определение цинка производят следующим образом.
Солянокислый раствор цинка из центрифужной пробирки переливают в стакан емкостью 30 — 50 мл и споласкивают пробирку в тот же стакан 2 — 3 мл воды. (Если осадок сернистого цинка растворялся на фильтре, то последний промывают 2 — 3 мл воды, собирая промывные воды и солянокислый раствор цинка в стакан на 30 — 50 мл.)
К солянокислому раствору цинка в стакан добавляют 1 — 2 капли 1% фенолфталеина и по каплям 10% раствор соды (Na2CO3) до ясной розовой окраски. Жидкость в стакане нагревают до кипения и кипятят в течение 5 — 10 минут, чтобы перевести хлопьевидный осадок цинка в кристаллический (ZnCO3).
Через 5 — 10 минут осадок углекислого цинка отфильтровывают количественно, переводя из стакана на небольшой беззольный фильтр, промывают 5 — 6 раз горячей дистиллированой водой, подсушивают, вместе с фильтром переносят во взвешенный тигель и прокаливают. После выдержки в эксикаторе тигель с окисью цинка взвешивают. Вес ZnO умножают на коэффициент 0,8033 и получают вес металлического цинка в исследуемом растворе. Прокаленный остаток после охлаждения должен быть белого цвета. Бурая окраска указывает на примесь железа. В последнем случае цинк должен быть переосажден.
Содержание цинка (X) в мг/л вычисляют по формуле:
где а — вес окиси цинка, в мг; V — количество вытяжки, взятое для определения свинца, меди и цинка, в мл; 0,8033 — коэффициент пересчета окиси цинка на металлический цинк.
ОПРЕДЕЛЕНИЕ ТИТАНА
Качественное определение титана
Метод основан на образовании комплексного соединения пероксидисульфотитановой кислоты — H2[TiO(H2O2)(SO4)2] <*> — желто-оранжевого цвета при добавлении перекиси водорода к сернокислому раствору титана:
H2[TiO(SO4)2] + H2O2 = H2[TiO(H2O2)(SO4)2].
<*> Пероксидисульфотитановую кислоту часто называют надтитановой кислотой.
Реакция открывает до 5 — 10 гамма титана; она применяется для определения малых количеств титана — не более 0,1 мг в 1 мл раствора.
Мешающие элементы:
1. Железо, никель, хром, кобальт, которые своей окраской маскируют желто-оранжевую окраску пероксидисульфотитановой кислоты.
2. Ванадий, молибден, церий и отчасти хром — вследствие образования этими элементами с перекисью водорода окрашенных соединений.
3. Фториды, образуя с титаном комплексное соединение, значительно ослабляют желто-оранжевое окрашивание, образуемое титаном с перекисью водорода, вплоть до обесцвечивания в зависимости от количества фторидов.
4. Большое количество сульфатов щелочных металлов несколько ослабляет интенсивность желто-оранжевого цвета раствора и тем сильнее, чем меньше концентрация серной кислоты.
5. Большое количество фосфатов, лимонная кислота обесцвечивают раствор; винная кислота практически не влияет.
Реактивы
1. Стандартный раствор титана: 0,1663 г двуокиси титана (TiO2) сплавляют в платиновой чашке с 3 г смеси углекислых калия и натрия или с шестикратным количеством пиросернокислого калия (K2S2O7), выщелачивают 250 мл серной кислоты (1:2) и, после растворения солей, доливают раствор серной кислотой той же концентрации до 1 л (1 мл раствора соответствует 0,1 мг титана). Титр полученного раствора можно проверить следующим способом: 20 мл раствора осаждают аммиаком, осадок ортотитановой кислоты промывают водой, прокаливают и взвешивают. Фактор пересчета полученной при прокаливании двуокиси титана (TiO2) на титан равен 0,5995.
2. Перекись водорода, 3% раствор, не содержащий фтора. Испытание перекиси водорода на фтор: к 50 мл перекиси водорода добавляют 0,1 г окиси магния и выпаривают до небольшого объема. Сконцентрированный раствор переносят на часовое стекло и выпаривают досуха. Сухой остаток смачивают концентрированной серной кислотой и дают стоять 2 — 3 часа. По истечении этого времени содержимое стекла смывают и стекло высушивают. На стекле не должно быть следов вытравливания.
3. Аммиак 10%.
4. Аммоний надсернокислый (персульфат аммония), 25% раствор.
5. Кислоты: азотная, концентрированная; серная, концентрированная; серная, 1:3; серная, 10% раствор; фосфорная, 10% раствор.
6. Магний, окись.
7. Натр едкий, 20% раствор.
8. Натр едкий, 1% раствор.
9. Натрий углекислый, 1% раствор.
Ход определения
50 мл исследуемой вытяжки выпаривают в фарфоровой чашке досуха, высушивают в сушильном шкафу и озоляют в муфельной печи (или на газовой горелке). Золу растворяют в нескольких миллилитрах (3 — 5) серной кислоты (1:3) при нагревании, добавляют 5 капель концентрированной HNO3 и выпаривают до появления паров серной кислоты.
К охлажденному сернокислому раствору осторожно при перемешивании добавляют 10 мл дистиллированной воды и, если нужно, центрифугируют, собирая раствор в маленький стаканчик (N 1). К прозрачному раствору прибавляют 3 мл 25%, свежеприготовленного раствора персульфата аммония, делают метку на стаканчике карандашом на уровне жидкости и кипятят в течение 10 минут, добавляя время от времени в стакан горячую воду (дистиллированную) до метки. При наличии хрома последний при этом окисляется до шестивалентного. Затем в еще горячий раствор добавляют маленькими порциями при взбалтывании и нагревании раствора 20% раствор едкого натра до нейтральной реакции и его избыток — 2 — 3 мл. При этом титан и железо выпадают в осадок <*>. Осадок отделяют от раствора центрифугированием, стаканчик и осадок в пробирке промывают 1% горячим раствором едкого натра. Для этого в стаканчик наливают 3 мл раствора едкого натра (1%), споласкивают им стенки стаканчика и переносят в центрифужную пробирку с осадком. Содержимое пробирки взбалтывают таким образом, чтобы осадок равномерно распределился в жидкости. Затем стаканчик (N 1) еще раз промывают 2 мл горячего раствора едкого натра, перенося его в ту же центрифужную пробирку, перемешивают содержимое и центрифугируют.
<*> В присутствии железа создаются наилучшие условия для полного осаждения титана. Если в исследуемом растворе железо отсутствует, то целесообразно ввести его в испытуемый раствор в виде следов.
Для лучшего отделения мешающих элементов проводят переосаждение. Для этого осадок, собранный в центрифужную пробирку, растворяют в возможно малом количестве горячего 10% раствора серной кислоты, переводят раствор количественно в стаканчик (N 1), в котором проводилось первое осаждение титана и железа. Пробирку споласкивают небольшим количеством дистиллированной воды (2 — 3 мл) и переносят раствор в стаканчик (N 1). Затем в стаканчик добавляют 3 мл раствора персульфата аммония и водный раствор аммиака в избытке (раствор должен сильно пахнуть аммиаком), накрывают стаканчик часовым стеклом и кипятят раствор в течение 10 — 15 минут. При кипячении в стаканчик время от времени добавляют горячий раствор аммиака до метки, сделанной перед кипячением на стенке стаканчика. Охлаждают содержимое стаканчика и отделяют осадок от раствора центрифугированием. Стаканчик, в котором проводилось переосаждение, и осадок в центрифужной пробирке промывают горячим 1% раствором углекислого натрия, как указано выше. Осадок в центрифужной пробирке растворяют в 1 мл 10% раствора серной кислоты и переносят раствор в стаканчик (N 1), в котором проводилось переосаждение. Пробирку дважды промывают дистиллированной водой (расходуя на это 5 мл) и присоединяют промывные воды к раствору в стаканчике (N 1). Далее к содержимому стаканчика добавляют 1 — 3 мл фосфорной кислоты (10%) до исчезновения желтого окрашивания раствора <*>, обусловленного наличием в растворе железа. К бесцветному раствору добавляют 3 — 5 мл 3% раствора перекиси водорода (свежеприготовленного). При наличии титана раствор окрашивается в оранжево-желтый цвет (при малых количествах титана — желтый или светло-желтый).
<*> При этом железо образует бесцветный комплекс [Fe(PO4)]2.
Полученный раствор может быть использован, если это необходимо, для количественного определения титана.
Количественное определение титана
В стаканчик N 2, одинаковый по объему со стаканчиком N 1, в котором находится исследуемый на титан раствор, вносят то же количество дистиллированной воды и реактивов, что и для испытуемого раствора. Далее в стаканчик N 2 добавляют при постоянном помешивании из микробюретки (или градуированной на сотые доли миллилитра пипетки) стандартный раствор титана до тех пор, пока окраска жидкости в обоих стаканчиках станет приблизительно одинаковой. Затем содержимое каждого стаканчика количественно с помощью дистиллированной воды переводят в мерные колбы на 25 мл (соответственно колбы N 1 и 2), доводят содержимое каждой колбы до метки и хорошо перемешивают. Сравнение окрасок растворов проводят в цилиндрах Генера. Содержание титана в мг/л вычисляют по формуле:
| X = | а x 0,1 x h1 x 1000 | , |
| h x 50 |
где а — количество стандартного раствора титана, добавленное в стаканчик N 2, в мг; 0,1 — количество титана в 1 мл стандартного раствора, в мг; h1 — высота столба стандартного раствора (из колбы N 2) в цилиндре Генера; h — высота столба исследуемого раствора (из колбы N 1) в цилиндре Генера.
ОПРЕДЕЛЕНИЕ ФТОРА
Метод основан на обесцвечивании розового торий-ализаринового лака ионами фтора:
| (C14H7O4SO3)4Th + 6NaF + 4HCl -> |
| розового цвета |
| -> Na2ThF6 + 4C14H7O4SO3H + 4NaCl |
| желтого цвета. |
Освободившаяся ализаринсульфокислота имеет желтый цвет. После того как весь фтор будет связан, желтый цвет раствора от добавления малейших количеств азотнокислого тория изменяется в розовый вследствие образования торий-ализаринового лака.
Чувствительность метода 0,01 мг в 1 л.
Реактивы
1. Ализариновый красный С (ализарин S; 1,2-диоксиантрахинон-3-сульфокислота, натриевая соль, C14H7O4SO3Na x H2O). 0,5 г препарата растворяют в 1 л воды (бидистиллята) — запасной раствор. Для работы готовят 0,0125% раствор (25 мл основного раствора разбавляют до 100 мл).
2. Кальций уксуснокислый. Вначале готовят углекислый кальций, свободный от фтора, с помощью следующих реактивов:
а) раствор углекислого аммония. Растворяют 110 г углекислого аммония в небольшом количестве воды, добавляют 55 мл аммиака (уд. в. 0,880) и разбавляют до 600 мл;
б) 200 г сухого хлористого кальция, ч.д.а., растворяют в 600 мл теплой дистиллированной воды. В этот раствор вливают 20 мл углекислого аммония (реактив А), смесь нагревают до кипения, в течение нескольких минут дают осадку осесть, затем раствор фильтруют через пористую воронку с колбой для отсасывания или через обычную воронку, содержащую небольшой комочек ваты, а осадок, содержащий фтор, выбрасывают. Осаждение и отделение осадка, как это указано выше, повторяют 3 раза. К прозрачному фильтрату, не содержащему фтора, добавляют оставшееся количество углекислого аммония (реактив А), хорошо размешивают, дают осадку осесть, затем раствор отфильтровывают. Осадок промывают горячей водой до отрицательной реакции на хлориды и сушат при 100 град. C.
Из полученного углекислого кальция готовят приблизительно нормальный раствор уксуснокислого кальция следующим способом: 10 г углекислого кальция переносят в колбу объемом около 500 мл, добавляют 12 мл ледяной уксусной кислоты (или соответствующее количество уксусной кислоты иной концентрации), осторожно перемешивают и, чтобы началась реакция, осторожно добавляют небольшое количество воды (бидистиллята). Когда реакция прекратится, добавляют новую порцию воды и время от времени взбалтывают. Всего небольшими порциями добавляют около 150 мл воды (бидистиллята). Затем раствор кипятят до растворения осадка и проверяют реакцию на лакмус.
Если реакция раствора нейтральная или слабощелочная, его переливают в мерную колбу на 200 мл, охлаждают и доводят водой до метки. Если реакция раствора кислая, добавляют на кончике ножа углекислого кальция и жидкость снова кипятят до тех пор, пока реакция раствора не будет нейтральной или слабощелочной на лакмус.
3. Натр едкий, 0,05-н (0,1-н раствор. NaOH разбавляют равным количеством воды).
4. Натр едкий, 10% раствор.
5. Натрий фтористый (стандартный раствор). 100 — 150 г товарного препарата переносят в химический стакан, добавляют около 200 — 250 мл дистиллированной воды, хорошо перемешивают и фильтруют в фарфоровую чашку. Затем раствор упаривают на водяной бане до образования на его поверхности твердой корочки, которую снимают на фильтровальную бумагу, отжимают ее между листами, измельчают стеклянной палочкой и высушивают на воздухе.
0,2210 г чистой сухой соли растворяют в 100 мл бидистиллята (1 мл = 1 мг F). Раствор хранят в пропарафинированной склянке (запасной раствор). Для работы 1 мл запасного раствора разбавляют до 100 мл (1 мл = 0,01 мг F).
6. Песок кремневый чистый, свободный от фтора. Чистый кремневый песок обливают в платиновой чашке концентрированной серной кислотой и осторожно нагревают до исчезновения белых паров, после чего охлаждают и промывают водой (бидистиллятом).
7. Серебро сернокислое в кристаллах.
8. Серная кислота, концентрированная, кипятят до густых белых паров.
9. Серная кислота, 1:1. Реактив 8 разбавляют 1:1 бидистиллятом.
10. Соляная кислота, 1-н раствор.
11. Торий азотнокислый [Th(NO3)4 x 4H2O], 0,01-н раствор. Содержание азотнокислого тория в препарате проверяют путем прокаливания (переводят в ThO2). Если количество азотнокислого тория в препарате составляет, например, 99,4%, то берут навеску, равную 1,3883 г, и растворяют в 1 л бидистиллята (0,01-н раствор). Для работы 100 мл 0,01-н раствора разбавляют до 1 л (0,001-н раствор).
12. Реактив 12. Берут 16 мл 0,01-н раствора азотнокислого тория (см. реактив 11) и 37,5 мл нормальной соляной кислоты, разбавляют до 1 л.
Аппаратура
Перегонная колба объемом на 50 мл, парообразователь — стеклянная круглодонная колба объемом около 300 мл, холодильник.
Перегонная колба (рис. 11) закрывается резиновой пробкой с двумя отверстиями, через которые проходят термометр на 150 град. и Г-образная стеклянная трубка; термометр и трубка доходят почти до дна колбы. Наружный конец изогнутой трубки соединяется с парообразователем. Боковой отросток перегонной колбы по всей длине имеет вдавления, образующие внутри «елочку». Конец отростка соединяется с холодильником с помощью шлифованной пробки (можно пользоваться и резиновой пробкой).
Парообразователь закрывают резиновой или корковой пробкой с двумя трубками: короткой для отвода пара и длинной, доходящей почти до дна колбы и поднимающейся вверх приблизительно на 1 м (для уравнивания давления). Последняя трубка не должна быть узкой, чтобы не создавать давления в колбе выше атмосферного. Пар получают из бидистиллята, к которому добавляют 10% раствор едкого натра до щелочной реакции.
Минерализация органического вещества
200 мл вытяжки выпаривают частями следующим способом: 30 — 40 мл раствора переносят в фарфоровую чашку, добавляют 10 мл уксуснокислого кальция (реактив 2) и хорошо перемешивают. Затем добавляют 10% раствор едкого натра до слабощелочной реакции на лакмус и выпаривают на водяной бане досуха. После того как выпарится первая порция раствора, добавляют следующую, раствор снова нейтрализуют едким натром, доводя реакцию до слабощелочной, и выпаривают досуха. После того как все 200 мл вытяжки будут выпарены, чашку с сухим остатком помещают в холодный муфель у самой дверцы и нагревают его, оставляя дверцу открытой. По мере нагревания муфеля, еще задолго до появления слабого покраснения, содержимое чашки начинает обугливаться и сгорает. Как только появится заметное покраснение муфеля, его выключают. Обычно к этому времени весь уголь сгорает; остаются несгоревшими лишь частицы угля на стенках чашки. Частицы угля отделяют от стенок чашки с помощью стеклянной хорошо оплавленной палочки, золу перемешивают и снова ставят в муфель при тех же условиях. Обычно озоление заканчивается через 2 — 3 часа. Зола — белого или серовато-белого цвета.
Отделение фтора от элементов, мешающих его определению, дистилляцией
В перегонную колбу помещают длинную стеклянную трубку, имеющую вверху расширение в виде воронки. Нижний конец трубки должен находиться в колбе ниже ее горлышка. Вносят в колбу небольшое количество кремневого песка и сернокислого серебра в количестве, достаточном для полного связывания хлоридов, если таковые имеются в исследуемом растворе <*>. Далее в колбу переносят золу исследуемой вытяжки. Оставшиеся в чашке частицы золы смывают 2,5 мл воды (бидистиллята) и 2,5 мл воды, содержащей каплю серной кислоты (1:1). После этого чашку и трубку обмывают 10 мл серной кислоты, разведенной 1:1, беря ее небольшими порциями с тем, чтобы при добавлении кислоты к золе, находящейся в колбе, не было бурного выделения углекислоты, которое может быть причиной потери фтора. Не касаясь стенок колбы, осторожно вынимают стеклянную трубку из колбы. Колбу закрывают пробкой, помещают на асбестовую сетку, имеющую посредине отверстие, и соединяют с холодильником. Во избежание местного перегрева стенок перегонной колбы последнюю обкладывают асбестом. Конец холодильника опускают в воду приемника — колориметрическую пробирку с делением на 10 мл, содержащую 5 мл воды (бидистиллята). Перегонную колбу соединяют с парообразователем, нагретым к этому времени до кипения, и нагревают до 135 град. Когда в холодильнике появится первая капелька жидкости (при 100 — 105 град.), колориметрическую пробирку, в которую был опущен конец холодильника, заменяют мерной колбой на 100 мл, не содержащей воды. Отгонку ведут при температуре около 135 +/- 3 град.
<*> Количество хлоридов в исследуемом растворе определяют общепринятыми методами.
После того как будет собрано 100 мл дистиллята, мерную колбу заменяют колориметрической пробиркой с делением на 10 мл. Когда в пробирке соберется 10 мл дистиллята, ее заменяют другой такой же пробиркой. Во все последующие приемники собирают по 10 мл отгона.
Продолжают отгонку до тех пор, пока на титрование последней фракции отгона пойдет 0,025 — 0,015 мл 0,001-н раствора азотнокислого тория (см. «Определение фтора в дистилляте»).
Чтобы убедиться в том, что минеральные кислоты — соляная кислота при недостаточном количестве добавленного сернокислого серебра и серная кислота при несоблюдении описанных условий дистилляции — отсутствуют в дистилляторе, раствор в мерной колбе объемом 100 мл хорошо перемешивают, переносят каплю на предметное стекло и к ней добавляют каплю метилоранжа. Реакция должна быть нейтральной. В случае кислой реакции подготовку образца к определению фтора повторяют, беря новую порцию вытяжки из пластмассы или другого исследуемого материала.
Определение фтора в дистилляте
10 мл дистиллята из мерной колбы (на 100 мл) переносят в колориметрическую пробирку с делениями на 10 мл, добавляют 0,2 мл раствора ализарина (реактив 1) и по каплям — едкий натр (реактив 3) до фиолетово-розового окрашивания.
Обычно на нейтрализацию идет 1 — 2 капли 0,05-н раствора щелочи.
Далее добавляют 0,5 мл реактива 12 и титруют 0,001-н раствором азотнокислого тория, сравнивая окраску с контролем, который в колориметрической пробирке такого же диаметра и стекла содержит 10 мл бидистиллята, 0,2 мл раствора ализарина, 1 каплю едкого натра (0,05-н) и 0,5 мл реактива 12. Цвет контрольного раствора очень слабо-розовый.
Титрование проводят осторожно, добавляя раствор азотнокислого тория из микробюретки с делениями на 0,01 мл до одинаковой окраски испытуемого и контрольного раствора. Сравнение окрасок производят на белом фоне (например, на фильтровальной бумаге), рассматривая раствор через весь столб жидкости. Для того чтобы лучше заметить конец титрования, хорошо иметь еще одну пробирку, содержащую 10 мл бидистиллята, 0,2 гамма фтора, все необходимые реактивы, и к концу титрования рассматривать все 3 пробирки.
Все дистилляты, собранные в колориметрические пробирки, титруют так, как указано выше.
Так как реакция между азотнокислым торием и фтором идет не точно по уравнению, соотношение между 0,001-н раствором азотнокислого тория и количеством фтора устанавливают эмпирически, путем титрования различных количеств стандартного раствора фтора азотнокислым торием. Из полученных величин составляют таблицу для вычисления фтора по расходу 0,001-н раствора азотнокислого тория.
Титрование рекомендуется. При составлении таблицы начинать с 0,2 гамма фтора с интервалами в 0,2 гамма, т.е. брать 0,2; 0,4; 0,6; 0,8; 1 гамма и т.д., пользуясь при этом микробюреткой с делениями на 0,01 мл. Для каждой концентрации необходимо не менее трех титрований при хорошо сходящихся результатах.
Все применяемые растворы и реактивы должны быть проверены на содержание фтора с помощью контрольного опыта.
Пользуясь составленной таблицей, находят количество фтора для каждой фракции дистиллята. Полученные величины складывают, вычитают количество фтора, содержащееся в реактивах, и пересчитывают на 1 л раствора.
ОПРЕДЕЛЕНИЕ ХРОМА
Метод основан на окислении дифенилкарбазида бихроматом с образованием растворимого в условиях метода соединения красно-фиолетового цвета. Строение образующегося соединения неизвестно.
Чувствительность метода 0,15 гамма хрома.
Реактивы
1. Серная кислота, 1:3 и 1:9.
2. Азотная кислота, уд. в. 1,4.
3. Азотнокислое серебро, 0,6% раствор.
4. Персульфат аммония, 15% свежеприготовленный раствор.
5. Натрий хлористый, 0,2% раствор.
6. Натрий углекислый, 7,5% раствор.
7. Дифенилкарбазид, свежеприготовленный: 0,1 г дифенилкарбазида растворяют в 10 мл ледяной уксусной кислоты, к раствору добавляют 90 мл чистого спирта.
8. Двухромовокислый калий, 0,001-н раствор.
Чистый перекристаллизованный и высушенный при 130 град. C двухромовокислый калий в количестве 0,045 г растворяют в дистиллированной воде в литровой колбе, объем доводят до метки и хорошо перемешивают. 1 мл раствора соответствует 0,015 мг хрома.
Ход определения
50 — 100 мл вытяжки выпаривают в фарфоровой чашке на водяной бане досуха; полученный остаток осторожно озоляют. После охлаждения вводят в чашку 5 мл раствора серной кислоты (1:9), помещают чашечку на кипящую водяную баню и после полного растворения остатка содержимое чашки переводят вместе с промывной водой, которой ополаскивают чашку, в коническую колбу на 100 мл, прибавляют 8 — 10 капель концентрированной азотной кислоты, после чего кипятят жидкость до появления белых паров серной кислоты. По охлаждении к жидкости осторожно прибавляют 20 мл дистиллированной воды, 2 мл раствора 0,6% азотнокислого серебра и 2 мл раствора 15% персульфата аммония и кипятят до полного разрушения персульфата аммония (около 30 минут).
При этом в присутствии хрома раствор окрашивается в желтый или желтоватый цвет, а в присутствии марганца — в малиновый или слабо-малиновый.
Для разрушения марганцевой кислоты к раствору добавляют 4 мл раствора хлористого натрия и кипятят жидкость в течение 5 минут на асбестовой сетке. Если при этом малиновый цвет жидкости не исчезает, добавляют еще 4 мл раствора хлористого натрия и снова кипятят в течение 5 минут.
Далее колбу снимают с асбестовой сетки, добавляют 10 мл холодной дистиллированной воды и тотчас охлаждают под краном или помещают в сосуд со льдом. К охлажденному таким образом раствору осторожно прибавляют приблизительно 8 — 10 мл 7,5% раствора углекислого натрия для осаждения железа, избегая большого избытка углекислого натрия. После этого содержимое колбы переносят в мерную колбу на 50 мл, доводят ее дистиллированной водой до метки и тщательно перемешивают. После отстаивания жидкость фильтруют в сухую колбу через сухой складчатый фильтр, отбрасывая первую небольшую часть фильтрата.
20 мл прозрачного раствора переносят в мерную колбу на 25 мл, добавляют 5 мл едкого натра (1:10) и дистиллированную воду до метки колбы; хорошо перемешивают и помещают в кипящую водяную баню на 1 час и, если раствор мутный, фильтруют через сухой фильтр в сухую колбу Эрленмейера. К 2 — 3 мл прозрачного фильтрата добавляют избыток серной кислоты (1:3) и 1 — 2 мл 0,1% раствора дифенилкарбазида.
При наличии хрома раствор окрашивается в красно-фиолетовый цвет.
В случае необходимости проводят количественное определение хрома.
Для количественного определения хрома берут 20 мл фильтрата в мерную колбу емкостью 50 мл, добавляют по каплям серную кислоту (1:3) до кислой реакции на лакмус и прибавляют избыток последней — несколько капель. При помутнении жидкости после подкисления раствор фильтруют. К прозрачному раствору добавляют 5 мл раствора дифенилкарбазида, дистиллированной водой доводят объем в колбе до метки и оставляют в покое на несколько (5 — 10) минут.
В это время готовят стандартный раствор. В мерную колбу емкостью 50 мл наливают 20 мл дистиллированной воды, 5 мл серной кислоты (1:3), 5 мл раствора дифенилкарбазида и добавляют из микробюретки 0,001-н раствор двухромовокислого калия до получения окраски, близкой к окраске испытуемого раствора. Объем жидкости в колбе доводят дистиллированной водой до метки, хорошо перемешивают и оставляют в покое на 10 минут, после чего проводят сравнение окрасок испытуемого и стандартного растворов в цилиндрах Генера. Содержания хрома рассчитывают по следующей формуле:
| X = | а x h1 x 25 x 50 x 1000 | , |
| h x 20 x 20 x b |
где X — количество хрома в мг/л вытяжки; а — количество хрома, взятое для стандартного раствора; h1 — высота столба стандартного раствора; h — высота столба испытуемого раствора; b — количество вытяжки, взятое для анализа, в мл.
Для мойки посуды и аппаратуры, используемой при определении хрома, не должна применяться хромовая смесь.
Приложения
ДОПУСТИМЫЕ КОЛИЧЕСТВА МИГРАЦИИ (ДКМ) ВЕЩЕСТВ, ВЫДЕЛЯЮЩИХСЯ ИЗ ПОЛИМЕРНЫХ МАТЕРИАЛОВ В МОДЕЛЬНЫЕ СРЕДЫ, И МЕТОДЫ ИХ ОПРЕДЕЛЕНИЯ <*>
<*> Методы, указанные в данной таблице, являются официальными для определения перечисленных в ней веществ; другие методы их определения, изложенные в Инструкции, следует считать факультативными.
| Наименование вещества | ДКМ, в мг/л | Примечания |
| 1 | 2 | 3 |
| Бутилокситолуол (ионол, П-21) | 2,00 | |
| Гексаметилендиамин | 0,01 | Не должен обнаруживаться при определении: |
| 1. Колориметрическим методом с 2,4-динитрохлорбензолом. Чувствительность 1,25 мг/л. | ||
| 2. Колориметрическим методом по реакции с нингидрином после выделения гексаметилендиамина посредством бумажной хроматографии. Чувствительность 1 мг/л | ||
| Дибутилсебацинат | 4,00 | Колориметрический метод по реакции спиртов, полученных после гидролиза сложных эфиров с ароматическими альдегидами. Чувствительность 0,05 мг/л |
| Диоктилсебацинат | 4,00 | |
| Дибутилфталат | 0,25 | |
| Диизооктилфталат | 2,00 | Не должен обнаруживаться при определении по реакции образования фенолфталеина. Чувствительность метода 2 мг/л |
| Дифенилолпропан | 0,01 | Не должен обнаруживаться при определении колориметрическим методом по образованию нитрозосоединения при взаимодействии дифенилолпропана с азотистой кислотой. Чувствительность 0,02 мг/л |
| Е-капролактам | 0,60 | Не должен обнаруживаться при определении колориметрическим методом аммонийного азота по Несслеру после минерализации Е-капролактама |
| Малеиновый ангидрид | 0,50 | |
| Меламин | 1,00 | Не должен обнаруживаться микрореакциями |
| Метиловый спирт (метанол) | 1,00 | Колориметрический метод определения метилового спирта с хромотроповой кислотой. Чувствительность 0,25 мг/л |
| Метилметакрилат | 0,25 | — |
| Нитрил акриловой кислоты | 0,05 | Не должен обнаруживаться при определении колориметрическим методом по аммиаку с реактивом Несслера. Чувствительность метода 0,2 мг нитрила акриловой кислоты в 1 л |
| 2-окси-4-метоксибензофенон | 2,0 | — |
| П-23 (2,4,6-тритрет-бутилфенол) | 2,0 | — |
| ОП-10 | 1,0 | Колориметрический метод определения окрашенного производного октилфенола с м-нитробензальдегидом после гидролиза ОП-10. Чувствительность метода 0,2 мг/л |
| Полиэтиленполиамин | 0,01 | Не должен обнаруживаться при определении по триэтилентетрамину с эозином К. Чувствительность 1 мг/л |
| Стирол | 0,01 | 1. Не должен обнаруживаться при определении методом нитрования. Чувствительность метода 0,075 мг/л. |
| 2. Не должен обнаруживаться при определении его с формалинсерным реактивом. Чувствительность метода 0,05 мг/л. | ||
| 3. Не должен обнаруживаться при определении его спектрофотометрическим методом. Чувствительность метода 0,02 мг/л | ||
| Тинувин Р [беназол, 2(2′-окси-5-метилфенил)-бензотриазол] | Спектрофотометрический метод определения. Чувствительность 0,05 мг/л | |
| Тринонилфенилфосфит (полиград, фосфит Н.Ф) | 2,00 | |
| Уротропин | 2,00 | Колориметрический метод определения по формальдегиду с хромотроповой кислотой. Чувствительность метода 0,25 мг/л |
| Фенол | 0,001 | Не должен обнаруживаться: 1. Колориметрическим методом по реакции с 4-аминоантипирином. Чувствительность 0,1 мг фенола/л. 2. Колориметрическим методом по реакции с диазотированным п-нитроанилином. Чувствительность 0,1 мг/л |
| Формальдегид | 0,10 | Колориметрический метод определения по реакции с хромотроповой кислотой. Чувствительность метода 0,1 мг/л |
| Фталевый ангидрид | 0,20 | Не должен обнаруживаться при определении колориметрическим методом по реакции образования фенолфталеина. Чувствительность метода 2 мг/л |
| Эпихлоргидрин | 0,10 | Не должен обнаруживаться при определении колориметрическим методом по реакции с хромотроповой кислотой после окисления эпихлоргидрина до формальдегида. Чувствительность метода 0,25 мг/л |
| Медь | Не допускается | Не должна обнаруживаться по методу, изложенному выше |
| Мышьяк | Не допускается | Не должен обнаруживаться по методу, изложенному выше |
| Свинец | Не допускается | Не должен обнаруживаться по методу, изложенному выше |
| Титан | 0,10 | Не должен обнаруживаться при определении методами, изложенными выше |
| Фтор | 0,50 | Колориметрический торий-ализариновый метод. Чувствительность 0,01 мг/л |
| Хром | Не допускается | Не должен обнаруживаться по методу, изложенному выше |
| Цинк | Не допускается | Не должен обнаруживаться по методу, изложенному выше |
ПРЕДЕЛЬНО ДОПУСТИМЫЕ КОНЦЕНТРАЦИИ ВРЕДНЫХ ВЕЩЕСТВ В АТМОСФЕРНОМ ВОЗДУХЕ НАСЕЛЕННЫХ МЕСТ
| Наименование загрязняющих веществ | Предельно допустимые концентрации в мг/куб. м | |
| среднесуточная | максимальноразовая | |
| 1 | 2 | 3 |
| Ацетон | 0,35 | |
| Бензол | 0,80 | |
| Бензин (нефтяной, малосернистый в пересчете на углерод) | 1,5 | |
| Бензин сланцевый (в пересчете на углерод) | 0,05 | |
| Бутилацетат | 0,10 | |
| Бутиловый спирт | — | 0,3 |
| Винилацетат | 0,20 | |
| Гексаметилендиамин | 0,001 | |
| Дивинил | 1,0 | |
| Диметиланилин | — | 0,0055 |
| Изопропилбензол | 0,014 | |
| Изопропилбензола гидроперекись | 0,007 | |
| Капролактам (пары, аэрозоль) | 0,06 | |
| Малеиновый ангидрид (пары, аэрозоль) | 0,05 | |
| Метанол | 0,5 | |
| Метилакрилат | — | 0,01 |
| Метилмеркаптан | — | 9 x 10(-6) |
| Метилметакрилат | 0,1 | |
| Мышьяк (неорганические соединения, кроме мышьяковистого водорода, в пересчете на мышьяк) | 0,003 | |
| Пиридин | 0,08 | |
| Пропилен | 3,0 | |
| Пропиловый спирт | — | 0,3 |
| Сероуглерод | 0,01 | |
| Стирол | 0,003 | |
| Толуол | 0,6 | |
| Фенол | 0,01 | |
| Формальдегид | 0,012 | |
| Фталевый ангидрид (пары, аэрозоль) | — | 0,10 |
| Фтористые соединения (в пересчете на фтор): | ||
| газообразные соединения (HF, SiF4) | 0,005 | |
| хорошо растворимые неорганические фториды (NaF, Na2SiF6) | 0,01 | |
| плохо растворимые неорганические фториды (AlF3, Na3AlF6CaF2) | 0,03 | |
| при совместном присутствии газообразного фтора и фторосолей | 0,01 | |
| Фурфурол | 0,05 | |
| Хлор | 0,03 | |
| Хром шестивалентный (в пересчете на Cr2O3) | 0,0015 | |
| Циклогексанол | 0,06 | |
| Циклогексанон | 0,04 | |
| Эпихлоргидрин | 0,2 | |
| Этилацетат | 0,1 | |
| Этилен | 3,0 |
ПЕРЕЧЕНЬ
ВЕЩЕСТВ, РАЗРЕШЕННЫХ ДЛЯ ВВЕДЕНИЯ В ПОЛИМЕРНЫЕ МАТЕРИАЛЫ, ПРЕДНАЗНАЧЕННЫЕ ДЛЯ КОНТАКТА С ПИЩЕВЫМИ ПРОДУКТАМИ
| Наименование вещества | ГОСТ, ТУ, МРТУ | Химическое название |
| 1 | 2 | 3 |
| Красители и пигменты | ||
| Двуокись титана | ГОСТ 9808-65 | Двуокись титана |
| Лак рубиновый СК | ГОСТ 7436-55 | Кальциевая соль 4-аминотолуидин-3-сульфокислоты и 2,3-оксинафтойной кислоты |
| Литопон | ГОСТ 907-53 | Эквимолекулярная смесь сернистого цинка и сернокислого бария с примесью небольших количеств окиси цинка |
| Пигмент голубой фталоцианиновый | ГОСТ 6220-52 | Комплексная медная соль тетрабензотетра-азопорфирина |
| Пигмент железоокисный красный К | МРТУ 6-10-602-66 | Окись железа |
| Ультрамарин «УС» | ОСТ НКТП 31-60 | Алюмосиликат натрия, содержащий серу |
| Стабилизаторы и антиоксиданты | ||
| Алкофен БП, П-21 | МРТУ 12-Н-49-63 | 2,6-ди-трет-бутил-4-метил-фенол |
| Алкофен ДИП | 2,6-диизоборнил-4-метилфенол | |
| Бензон ОА | ВТУ 16-64 | 2-окси-4-алк(C7-C9) оксибензофенон |
| Бензон ОО | 2-окси-4-октоксибензофенон | |
| Бисалкофен БП | СТУ 36-13-32-64 | Бис-(5-метил-3-трет-бутил-2-оксифенил)-метан |
| Дилаурил-бета, бета’-тиодипропионат | ||
| Тиоалкофен БП | ВТУ 16-64 | Бис-(5-метил-3-трет-бутил-2-оксифенил)-моносульфид |
| Тиоалкофен МБП | ВТУ 7-63 | Бис-(5-метил-3-метилбензил-2-оксифенил)-моносульфид |
| Стеарат К | ТУ 51-54 | Стеарат кальция (в качестве смазки) |
| Стеарат Ц | ТУ МППТ 16-53 | Стеарат цинка |
ФОРМА ПРОТОКОЛОВ АНАЛИЗА ОБРАЗЦОВ ПОСУДЫ, ТАРЫ И ДРУГИХ ИЗДЕЛИЙ
МИНИСТЕРСТВО ЗДРАВООХРАНЕНИЯ СССР
НАЧАЛЬНИКУ ГЛАВНОГО САНЭПИДУПРАВЛЕНИЯ
На Ваш N ________ от ________ 197_ г.
АНАЛИЗ N 21
СТАКАНЫ ИЗ МЕЛАЛИТА (1 ОБРАЗЕЦ)
Образец доставлен завернутым в бумагу, перевязанным шпагатом, концы которого скреплены металлической пломбой с неразборчивым оттиском. Согласно письму (указать организацию) за подписью ______ ______________ от «___» _______________ 197_ г. N _______________, в рецептуру мелалита входят следующие ингредиенты:
1. Меламин.
2. Формальдегид.
3. Целлюлоза сульфитная.
4. Двуокись титана.
5. Пигмент голубой фталоцианиновый.
Стаканы предназначены для контакта с горячими и холодными напитками.
Образцы представляют собой стаканы в количестве 5 штук, емкостью около 300 мл, с внутренней площадью 250 кв. см. Цвет образца — серовато-голубой. Наружная и внутренняя поверхность стаканов блестящая, гладкая. На дне стакана имеется оттиск: «КПЗ, 492, 3, 20, Iе, цена 15 к.».
Запах без особенностей.
Стаканы промывали водопроводной, а затем дистиллированной водой и наполняли модельными растворами, нагретыми до 80 град.: 1) дистиллированной водой; 2) 1% раствором уксусной кислоты и оставляли при комнатной температуре на 2 часа.
После вышеуказанной экспозиции растворы переливали в стеклянные колбы с притертыми пробками и подвергали их органолептическому и химическому исследованиям. Образец после обработки подвергался осмотру.
Поверхность и цвет образца без особенностей по сравнению с контролем.
Вытяжки бесцветные, прозрачные, отмечается наличие постороннего ароматического запаха интенсивностью 4 балла. Вкус водной вытяжки посторонний, неприятный по сравнению с контролем.
При химическом исследовании получены следующие результаты.
В водной вытяжке обнаружены органические вещества, требующие для своего окисления 14,9 мг O2 на л (бихроматным методом).
Формальдегид в уксуснокислой вытяжке по реакции с хромотроповой кислотой обнаружен в количестве 16,1 мг/л; площадь образца, контактировавшая с модельным раствором, равна 250 кв. см; количество раствора, взятое для обработки вышеуказанной площади, 300 мл.
Анализ проводил (подпись)
ЗАКЛЮЧЕНИЕ
Исследованный образец стаканов, изготовленных из мелалита по нижеуказанной рецептуре, предназначенный для контакта с горячими и холодными напитками, вследствие изменения органолептических свойств вытяжек (посторонний запах интенсивностью 4 балла, посторонний неприятный привкус) и перехода в водную вытяжку большого количества органических веществ (окисляемость 14,9 мг O2/л), а также вследствие перехода в уксуснокислую вытяжку формальдегида в количестве 16,1 мг/л в условиях опыта признается неудовлетворительным и непригодным для использования по назначению.
Рецептура мелалита: меламин, формальдегид, целлюлоза сульфатная, двуокись титана, пигмент голубой фталоцианиновый.
Подпись руководителя учреждения
МИНИСТЕРСТВО ЗДРАВООХРАНЕНИЯ СССР
ЗАМЕСТИТЕЛЮ ГЛАВНОГО САНИТАРНОГО ВРАЧА СССР Д.Н. ЛОРАНСКОМУ
На Ваш N ________ от «__» _________ 197_ г.
Анализ N 45-46
Образцов древесины, обработанных клеями «МХ-4» и «К-17».
Образцы доставлены завернутыми в бумагу, неопечатанными.
В сопроводительном письме (наименование учреждения) за N _____ от «__» ___________ 197_ г. указано, что карбамидные клеи «МХ-4» и «К-17» предназначены для заделки дефектов древесины, идущей для изготовления бочек под тузлучные рыбопродукты.
Согласно представленной рецептуре в состав клеев входят следующие ингредиенты.
Клей «МХ-4»
1. Смола «М-4».
2. 50% раствор молочной кислоты.
Рецептура смолы «М-4»
1. Мочевина.
2. Формалин, 37% раствор.
3. Едкий натрий, 40% раствор.
4. Уротропин.
5. Хлористый цинк, 50% раствор.
Клей «К-17»
1. Смола «МФ-17».
2. 15% раствор щавелевой кислоты.
Рецептура смолы «МФ-17»
1. Мочевина.
2. Формалин, 37% раствор.
3. Аммиачная вода, 25% раствор.
4. Диэтиленгликоль.
Образец N 1 представляет собой 10 штук деревянных пластин размером 9,5 x 8 см. Пластины, включая и торцы, покрыты клеем «МХ-4». Поверхность пластин, обработанная клеем, — гладкая, блестящая, местами отмечаются трещины. Запах образца — слабый, неприятный, ароматический, интенсивностью 1 балл.
Образец N 2 представляет собой 10 штук деревянных пластин размером 9,5 x 8 см. Пластины, включая и торцы, покрыты клеем «К-17». Поверхность пластин, обработанная клеем, — гладкая, блестящая. На поверхности некоторых образцов, обработанной клеем, отмечаются трещины. Запах образца — слабый, ароматический, интенсивностью 1 балл.
Пластины промывали водопроводной и дистиллированной водой, затем по 2 экземпляра каждого образца помещали в стеклянные стаканы с 500 мл дистиллированной воды, нагретой до кипения, и выдерживали в течение 2 часов при комнатной температуре.
После вышеуказанной экспозиции клей «К-17» на некоторых участках поверхности исследованных пластин вздулся и отслоился.
Цвет образца до обработки — светло-желтый, после соответствующей обработки водой — молочно-белый.
Полученные водные вытяжки после вышеуказанной обработки каждого образца древесины подвергались органолептическому исследованию. Результаты органолептического исследования вытяжек представлены в нижеследующей таблице:
| Наименование клея | Водная вытяжка |
| «МХ-4» | Вытяжка опалесцирует. Отмечается наличие хлопьевидных взвешенных частиц, оседающих при стоянии на дно. Запах вытяжки по сравнению с контролем резкий, посторонний, ароматический, интенсивностью 5 баллов |
| «К-17» | То же |
Анализ производил (подпись)
ЗАКЛЮЧЕНИЕ
Исследованные образцы древесины, обработанные карбамидным клеем «МХ-4» и «К-17», предназначенные для заделки дефектов древесины, идущей на изготовление бочек под тузлучные рыбопродукты, признаются неудовлетворительными и непригодными для использования по назначению вследствие наличия опалесценции, осадка в вытяжках и резкого, постороннего, ароматического запаха интенсивностью 5 баллов, а также вследствие плохой адгезии клея «К-17» к древесине.
Подпись руководителя учреждения
ДЕГУСТАЦИОННАЯ КАРТА
| Фамилия, имя, отчество | |||
| Дата анализа | |||
| N растворов, не отличающихся от контрольного | |||
| по запаху | по привкусу | ||
| N растворов, отличающихся от контрольного | |||
| по запаху | по привкусу | ||
| 1. Характер запаха исследуемого раствора (фенольный, ароматический посторонний, неопределенный и т.д.) | |||
| 2. Интенсивность запаха исследуемых растворов в баллах: |
| N растворов | Запах в баллах |
| 1 | |
| 2 | |
| 3 |
3. Характер привкуса исследуемого раствора (горьковатый, щиплющий, нефтепродуктов, посторонний, неопределенный и т.д.)
4. Интенсивность привкуса исследуемого раствора (слабый привкус, ясно выраженный, сильный)
ПЕРЕЧЕНЬ
СИНТЕТИЧЕСКИХ МАТЕРИАЛОВ, РАЗРЕШЕННЫХ ОРГАНАМИ ЗДРАВООХРАНЕНИЯ ДЛЯ ИСПОЛЬЗОВАНИЯ В ПИЩЕВОЙ ПРОМЫШЛЕННОСТИ <*>
<*> При условии соответствия изделий гигиеническим требованиям и регламентам, приведенным в Инструкции.
| N п/п | Наименование материала | ГОСТ или ТУ | Назначение | Рецептура | Дата и номер разрешения | Кем выдано разрешение |
| 1 | 2 | 3 | 4 | 5 | 6 | 7 |
| Отечественные материалы | ||||||
| 1 | Алюминиевые колпачки с картонной прокладкой, покрытой нелакированным целлофаном | Для укупорки бутылок с пищевым уксусом | 08с/Б-7-378 07.07.67 | Минздрав РСФСР | ||
| 2 | Алюминиевые пластины, покрытые оксидной пленкой, лакированной лаком ЭП-527 | Для изготовления испарителей холодильных агрегатов | См. рецептуру лака ЭП-527 | 126-5/363-3 01.08.68 | Минздрав СССР | |
| 3 | Барий сернокислый чистый ГОСТ 3158-65 | Компонент в рецептуре уплотнителей к кронепробкам | 123-9-28 11.03.64 | Минздрав СССР | ||
| 4 | Блок-полимер | Для укупорки сухих и крепленых вин крепостью не более 20 град. | Полиизобутилен П-200, полиэтилен н/д, белая сажа (SiO2) | 06с-3А/162 24.01.64 | Минздрав Молдавской ССР | |
| 5 | Бронза марки ОПС 5-5-5 | Для изготовления клапанов, предназначенных для слива молока из грузоприемных баков | 08с/Б-7-210 09.01.68 | Минздрав РСФСР | ||
| 6 | Бумага антиокислительная (с содержанием антиоксиданта не более 0,02%) | Для упаковки соленой сельди в ящики | Бутилокситолуол, бутилоксианизол, лимонная кислота | 3/3-73 24.01.64 | Минздрав Латвийской ССР | |
| 7 | Бумага парафинированная (парафином марки А и Б) | ГОСТ 9569-65 ГОСТ 784-53 ТУ УССР |
Для завертки только остывшего хлеба | 123-8/170-7 10.03.65 | Минздрав СССР | |
| 8 | Бумага ингибированная марки А и Б с полиэтиленовым покрытием | 13-0410-67 | Для упаковки мясорубок с целью предохранения от коррозии | Для упаковок марка А: бензоат натрия марка Б: бензоат аммония |
08с/Б-7-196 19.05.69 | Минздрав РСФСР |
| 9 | Бумага фольгаполиэтилен | Для контакта с пищевыми влажными продуктами при температуре 50 — 60 град. C, а также для упаковки пищевых концентратов, содержащих не более 15% жира | 126-14/624-3 27.06.69 | Минздрав СССР | ||
| 10 | Бумага, ламинированная полиэтиленом (П2070П) | Для упаковки сухих киселей (кислотность 1 — 1,3%, влажность 9 — 9,5%) | 08с/Б-7б2526 29.11.68 | Минздрав РСФСР | ||
| 11 | Бумага, пропитанная парафином марки «А» | ГОСТ 13577-68 | В качестве прокладки для рубленых бифштексов и упаковки мясных блоков, замораживаемых на мембранных скороморозильных аппаратах Шеффера | 125-54/ 1566-3 23.07.68 | Минздрав СССР | |
| 12 | Бутакрил | МРТУ 64-2-9-68 | Для изготовления вкладышей, предназначенных для облицовки роторов на кондитерских фабриках | Порошок — сополимер метилового эфира метакриловой кислоты и бутилового эфира метакриловой кислоты; жидкость — метиловый эфир метакриловой кислоты; ускоритель — диметиланилин; ингибитор — гидрохинон | 08с/Б-7-2805 09.06.69 | Минздрав РСФСР |
| 13 | Вазелиновое масло (вместо оливкового) | ГОСТ 3164-52 | Для смазывания колпачков из фольги, предназначенных для укупорки молочных бутылок | 08с/Б-7-961 27.09.66 | Минздрав РСФСР | |
| 14 | Восковой состав | Герметизация картонных коробочек, заполненных сухим пайком. Затаривание предварительно упакованных пищевых продуктов | На основе озокеритовых перезинов | 126-14/908-3 09.04.68 | Минздрав СССР | |
| 15 | Герметик | Для герметизации поперечного шва бумажного мешка | Церезин петролатумный ГОСТ 11057-64, полиизобутилен — высокомолекулярный П200 — ТУ МХП 1655-54, полиизобутилен низкомолекулярный П20 — ТУ МХП 1761-54р, полиэтилен низкой плотности, церезин синтетический ГОСТ 7658-55 | 123-14/ 1122-7 30.07.65 | Минздрав СССР | |
| 16 | Гидролизный спирт | Для растворения красителей при окрашивании пищевой алюминиевой фольги | 26.10.64 123-9/150 | Минздрав СССР | ||
| 17 | Глина бентонитовая, содержащая полиакриламид | Для отбелки пищевых масел | 08с/Б-7-1080 08.07.66 | Минздрав РСФСР | ||
| 18 | Дибуг (2,5-дитретбутилгидрохинон) | Применять в качестве антиоксидантов (стабилизаторов) в рецептурах резин и пластмасс, соприкасающихся с пищевыми продуктами, кроме резин, соприкасающихся с жирами | 08с/Б-7-1918 30.12.64 | Минздрав РСФСР | ||
| 19 | Дитаг (2,5-дитретамилгидрохинон) | То же | -« | -« | ||
| 20 | Дихлорэтан | ГОСТ 1942-63 | Для склеивания ящиков из оргстекла | 123-10-55 15.07.58 | Минздрав СССР | |
| 21 | Дихлордиметилгидантоин | Дезинфекция технологического оборудования консервных предприятий | 123-14/959-7 11.07.69 | Минздрав СССР | ||
| 22 | Доски из древесностружечных плит, изготовленных на основе мочевиноформальдегидных смол и покрытых защитной эмалью | Опытная партия досок для хранения сыров в пленке | Состав защитной эмали: эпоксидная смола ЭД-5, двуокись титана, полиэтиленполиамин, ацетон | 08с/Б-7-706 13.04.67 | Минздрав РСФСР | |
| 23 | Жесть белая, покрытая лаком ЭП-547 | Для консервной тары из лакированной жести | См. рецептуру лака ЭП-547 | 126-14/16663 03.12.68 | Минздрав СССР | |
| 24 | Жесть белая, покрытая лаком ЭП-527 | Для изготовления консервной тары (рыбных, мясных и плодоовощных консервов) | См. рецептуру лака ЭП-527 | 126-5/37-3 16.12.68 | Минздрав СССР | |
| 25 | Жесть хромированная, лакированная лаком ЭП-527 (банки, лакированные в два слоя, на внутреннюю поверхность корпусов цельноштампованных банок, второй слой наносят после изготовления банки, а крышки покрывают в один слой) | Для консервов мясных, рыбных и овощных | См. рецептуру лака ЭП-527 | 126-5/37-3 13.03.68 | Минздрав СССР | |
| 26 | Жесть черная и хромированная с покрытием лаком ЭП-547 | Для консервной тары | См. рецептуру лака ЭП-547 | 126-11/12133 26.12.68 | Минздрав СССР | |
| 27 | Жесть черная, покрытая лаком ЭП-527 | Изготовление крупной тары (банки N 14 и 15 под расфасовку повидла, джема, халвы, сухих овощей и фруктов) | 126-14/956-3 03.06.68 | Минздрав СССР | ||
| 28 | Жесть хромированная, покрытая лаком ЭП-527 (банка лакирована в 2 слоя, крышка — в один слой) | Затаривание консервов мясных, рыбных, овощных | 126-5/37-3 15.03.68 | Минздрав СССР | ||
| 29 | Ионол (бутилокситолуол) | В качестве стабилизатора пластмасс в количестве 0,02% | 123-9/137 01.10.64 | Минздрав СССР | ||
| 30 | Капролон | Детали машин, контактирующие с молоком и мясом | Е-капролактам ГОСТ 7850-63, натрий металлический ГОСТ 3273-63, натр едкий ГОСТ 4328-48, уксусный ангидрид ГОСТ 787-55 | 123-9/168 26.09.64 | Минздрав СССР | |
| 31 | Капрон | Шестерни, применяемые в кремосбивалках модели СК-1 при отсутствии контакта с пищевыми продуктами | Е-капролактам, уксусная кислота | 123-10/41 19.05.59 | Минздрав СССР | |
| 32 | Картон гофрированный, покрытый восковым составом | Для покрытия наружной поверхности лотков, предназначенных для затаривания плодоовощей | 126-14/12153 15.11.68 | Минздрав СССР | ||
| 33 | Картонные коробки с внутренним покрытием из термосваривающихся материалов (полиэтиленовая пленка П2070П) | Для упаковки сухих завтраков (кукурузные палочки, хлопья кукурузные и пшеничные, овсяные хлопья «Геркулес» и т.д.) | 08с/Б-7б2533 27.11.68 | Минздрав РСФСР | ||
| 34 | Картонные капсулы с наложением сверху алюминиевого колпачка | Для укупорки бутылок московской особой водки | 123-10/32 11.07.60 | Минздрав СССР | ||
| 35 | Картон склеенный, тарный | Для изготовления наружной тары, предназначенной для упаковки продуктов, имеющих герметическую упаковку | Сульфат небеленной целлюлозы, проклеенной канифольно-парафиновым или канифольным клеем и меламиновой смолой и склеенной поливинилацетатной эмульсией | 123-9/27 07.03.64 | Минздрав СССР | |
| 36 | Клей | ГОСТ 2067-47 | Для укупорочных композиционных пресс-пятачков | Пробковая крупка, раствор костного клея, глицерин, уротропин (ГОСТ 2267-41) | 123-10/17 17.02.60 | Минздрав СССР |
| 37 | Клей синтетический | Для оклейки стеклобанок и широкогорлых бутылок 58-2, при условии, что клей будут получать централизованно, но не готовить на месте, т.к. при изготовлении выделяются сильнопахнущие раздражающие пары | На основе мочевиноформальдегидной смолы МФ-РХП и мочевиноформальдегидно-фурфурольной смолы | 08с/Б-7-1163 18.07.66 | Минздрав РСФСР | |
| 38 | Синтетический клей (на основе мочевиноформальдегидной смолы и сульфитноспиртовой барды) <1> | Для оклейки стеклобанок и широкогорлых бутылок 58-2 на этикетировочных машинах | 08с/Б-7-464 22.04.67 | Минздрав РСФСР | ||
| 39 | Клей | Для склеивания фольги с подпергаментом | Крахмал, натр едкий, салициловая кислота, вода | 113-17/542 02.10.54 | Минздрав СССР | |
| 40 | Клей | Для приклеивания этикеток к таре с соблюдением требований гигиены труда при работе с клеем | Полиакриламид, натриевая соль карбоксиметилцеллюлозы, латекс СКС-30 марки ШР или Ш с добавлением клея, галерта | 123-9/42 08.04.64 | Минздрав СССР | |
| 41 | Клей | ГОСТ 8518-57 | Для наклейки этикеток | Концентрат сульфитноцеллюлозной барды марок КБЖ, КБТ, КБП в смеси с глицерином | 05-42/1041 01.10.64 | Минздрав СССР |
| 42 | Клей альбуминоказеиновый | Для покрытия и оклеивания фанеры, идущей на изготовление бочек под пищевые продукты (сухие, соленые и влажные, имеющие нейтральную или слабокислую реакцию) | Обесфеноленная фенолоформальдегидная смола, казеиновый клей | 113-17/173 28.07.53 | Минздрав СССР | |
| 43 | Клей декстриновый | Для наклеивания этикеток на чайные пачки | Крахмал, квасцы, сода каустическая, вода | 1455-31/8 15.12.65 | Минздрав Грузинской ССР | |
| 44 | Клей канифольно — петролатумный | Для подклейки прокладок к кроненпробкам с крышками СКК, предназначенными для укупорки безалкогольных и алкогольных напитков | Канифоль ГОСТ 797-64, петролатум марки ПК ГОСТ 4096-62 | 06с-3/147 22.01.64 | Минздрав Молдавской ССР | |
| 45 | Клей эпоксидный ЭКП | ГОСТ 10587-63 | В качестве связующего вещества при нанесении слоя кварцевого песка на внутреннюю поверхность деталей универсального комбайна УЭМ-1, предназначенного для чистки картофеля | Смола ЭД-6 ГОСТ 10587-63, дибутилфталат ГОСТ 2102-51, отвердитель — полиэтиленполиамин | 08с/Б-7-1687 22.07.65 | Минздрав РСФСР |
| 46 | Клей | ВТУ 102-58 | Для склеивания бумажных стаканов под напитки | Поливиниловый спирт, крахмальный клейстер | 08с/Б-7б-67 15.01.69 | Минздрав РСФСР |
| 47 | Клей для липкой ленты на полиэтиленовой основе | Для тары под пищевые продукты без контакта с ними | 126-14/325-3 15.03.68 | Минздрав СССР | ||
| 48 | Колпачки из фольги с пергаментной прокладкой | Для укупорки бутылок с растительным маслом | 08с/Б-7-277 27.01.64 | Минздрав РСФСР | ||
| 49 | Красители: естественные — кармин, шафран, индиго, сафлер, аннато, каратин; синтетические — индигокармин | В кондитерской, безалкогольной и др. отраслях пищевой промышленности | 08с/Б-7-821 18.05.65 | Минздрав РСФСР | ||
| 50 | Краситель желтозеленый М8, спирторастворимый | Для окраски алюминиевой фольги, предназначенной для упаковки кондитерских изделий | 123-8/90 17.10.57 | Минздрав СССР | ||
| 51 | Краситель оранжевый 4-Ж, спирторастворимый | Для окраски фольги, предназначенной для упаковки кондитерских изделий | C32H27O8N10S2CO | 123-10/57 04.07.58 | Минздрав СССР | |
| 52 | Краситель рубиновый спирторастворимый | Для окраски алюминиевой фольги | 123-8/56 05.06.57 | Минздрав СССР | ||
| 53 | Кремнийорганическая жидкость гидрофобизирующая ГКЖ-94 | ГОСТ 10834-64 | 1. Для покрытия стеклотары снаружи | Полиэтилгидросилаксан, растворитель — толуол или четыреххлористый углерод | 123-7/10 30.01.61 | Минздрав СССР |
| 2. Покрытие алюминиевых форм, предназначенных для изготовления ветчины и мясного хлеба | То же | 126-14/ 297-3 27.08.69 | Минздрав СССР | |||
| 3. Для обработки тесторазделочных линий в хлебопекарной промышленности | 123-10/31 10.03.60 | Минздрав СССР | ||||
| 54 | Крышки СКО-83 и СКО-58 из белой жести, покрытие лаком 3-30-59 | Для домашнего консервирования | Лак 3:30-59; 1) смола КФ-Э (ксиленол), фенол, формальдегид; 2) смола Э-05: смола Э-33 (дифенилолпропан, эпихлоргидрин, сульфат целлюлозы), дифенилолпропан; 3) смола Э-30Т или Э-30К (смола ГФ-019, масло льняное, масло тунговое, глицерин, фталевый ангидрид, каустическая сода; смола Э-40: дифенилолпропан, эпихлоргидрин, едкий натр; тетралин-тетрагидронафталин или ксилол) |
О8с/Б-7-2435 23.12.66 | Минздрав РСФСР | |
| 55 | Крышки СКО-83 из хромированной жести, покрытой лаком ЭП-527 в один слой (толщиной 5 — 6 микрон) | Укупорка мясных, овощных, фруктовых консервов в стеклотаре | См. рецептуру «Лак ЭП-527» | 126-8/127-3 13.03.68 | Минздрав СССР | |
| 56 | Лавсан (полиэтилентерефталатная пленка «О») | В качестве герметизирующего термостойкого материала для упаковки на длительное время | Диметилтерефталат, этиленгликоль | 123-14/868-7 01.04.65 | Минздрав СССР | |
| 57 | Лак бакелитовый А, Б | ГОСТ 901-56 | Для покрытия торгового оборудования | 113-17/66 19.03.52 | Минздрав СССР | |
| Лак N 18378 (В и С) | Для покрытия алюминия, применяемого в консервной промышленности | Ксиленолфенолформальдегидная смола, модифицированная жирными кислотами дегидратированного касторового масла. Циклогексанофенолформальдегидная смола, силиконовое масло; растворители: бутанол, этилцеллозольв | 123-9/13 31.01.64 | Минздрав СССР | ||
| 58 | Лак БФ-2 | Для покрытия рафинированных форм и лотков, применяемых в кондитерской промышленности, рабочих поверхностей аппаратуры и оборудования в винодельческой промышленности, хлебопекарного оборудования | 08с/Б-7-1390 25.08.66 | Минздрав РСФСР | ||
| 59 | Лак кислотостойкий 3-30-59 | ВТУ-ГИПИ4В | 1. Для внутреннего покрытия сиропных банок из алюминия марки Ае ГОСТ 3349-55 к автоматам АТ-100 для продажи газированной воды. Рекомендуется лак наносить на поверхность изготовленных банок | Ксиленолфенолформальдегидная смола КФ-Э: ксиленол, фенол, формальдегид; эпоксидная смола Э-05: эпоксидная смола Э-33 (дифенилолпропан, эпихлоргидрин, сульфат целлюлозы), дифенилолпропан, пиперидин | 08с/Б-7-1869 31.07.64 | Минздрав РСФСР |
| 2. Для покрытия металлических крышек | Алкидноэпоксидная смола Э-30Т (или Э-30К): полиэфирная смола ГФ-019 (масло льняное, масло тунговое, глицерин, фталевый ангидрид, каустическая сода), смола Э-40 (дифенилолпропан, эпихлоргидрин, едкий натр), тетралин (тетрагидронафталин) или ксилол | 08с/Б-7-3124 08.07.64 | Минздрав РСФСР | |||
| 3. Для покрытия внутренней поверхности консервных банок из белой жести горячего лужения | 08-113-18295 25.03.60 | Минздрав РСФСР | ||||
| 4. Для внутренней лакировки консервных банок | 08с/Б-7-625 17.05.66 | Минздрав РСФСР | ||||
| 60 | Лак СВМ-31 | В качестве покровного лака при глубокой и флексографической печати по алюминиевой фольге, применяемой для упаковки пищевых продуктов | На основе сополимера метилметакрилата с винил-нбутиловым эфиром с катализатором до 0,2% перекиси бензоила | 126-14/10843 01.07.69 | Минздрав СССР | |
| 61 | Лак 41-К | СТУ-30 9011-62 | Для лакировки белой жести под консервную тару | Копал «КГ» (крезолформальдегидная смола модифицированная), масло льняное оксидированное, масло касторовое дегидрированное, кобальтомарганцовый линолеат, скипидар | 09.02.62 | |
| 62 | Лак ЭП-527 | Для покрытия рам и пластин фильтрпрессов в винодельческой промышленности | 1. Смола ФПФ-1: паратретбутилфенол, фенол, формальдегид, едкий натр, соляная кислота, бутанол, ортофосфорная кислота | 08с/Б-7-1098 21.07.69 | Минздрав РСФСР | |
| 2. Смола эпоксидная Э-05: дифенилолпропан, эпихлоргидрин, едкий натр, карбоксиметилцеллюлоза (КМЦ) | ||||||
| 3. Смола алкидноэпоксидная Э-30Т (или Э-30К): масло касторовое, глицерин, фталиевый ангидрид, дифенилолпропан, эпихлоргидрин, тетралин (тетрагидронафталин) | ||||||
| 63 | Лак ЭП-527 для покрытия белой жести электролитического горячего лужения | Изготовление тары под рыбные консервы из тунца и других видов | 126-5/37-3 15.05.68 | Минздрав СССР | ||
| 64 | Лак ЭП-527 с подслоем оксидной пленки по рецептуре ванны N 1 | Для защиты от коррозии алюминиевых испарителей домашних холодильников | Лак ЭП-527. Ванна N 1: хромовый ангидрид, фторсиликат натрия | 126-11/567-3 30.06.69 | Минздрав СССР | |
| 65 | Лак ХС-76 с подслоем оксидной пленки по рецептуре ванны N 2 | Для защиты от коррозии алюминиевых испарителей домашних холодильников | Лак ХС-76. Ванна N 2: ортофосфорная кислота, хромовый ангидрид, фтористо-водородная кислота | 126-11/567-3 30.06.69 | Минздрав СССР | |
| 66 | Лак ЭП-547 с добавкой лака ФГ-9 <2> | Изготовление консервной тары | Смола ФПФ-1: паратретбутилфенол, фенол, формальдегид, едкий натр, соляная кислота, бутанол, ортофосфорная кислота | 126-14/483-3 11.07.69 | Минздрав СССР | |
| 2. Смола эпоксидная Э-05: дифенилолпропан, эпихлоргидрин, едкий натр, карбоксиметилцеллюлоза, ортофосфорная кислота | ||||||
| 3. Смола алкидносиликоновая ФГ-9 | ||||||
| 67 | Лак цветной | В пищевой промышленности для нанесения на внешнюю поверхность фольги | На основе ВЛ-564, НЦ-562 с использованием пигментов двуокиси титана, сажи газовой гранулированной, голубого фталоцианинового, лака рубинового СК | 126-5/410-3 04.10.68 | Минздрав СССР | |
| 68 | Лак ЯК-1 | ТУ 2079-49 | Покрытие деталей молочной аппаратуры | 126-14/334-3 12.02.68 | Минздрав СССР |
| 69 | Латекс СВХ-1 | 3478-52 | Для покрытия бумажной тары, для упаковки молочных продуктов | СВХ-1: продукт полимеризации винилиденхлорида и хлористого винила в присутствии персульфата аммония и никеля (натриевая соль дибутилнафталинсульфокислоты) | 123-9/165 02.11.64 | Минздрав СССР |
| 70 | Латекс СКС-65ГП | ГОСТ 10564-63 | Для склеивания картонной тары, предназначенной для упаковки только затаренных продуктов | 08с/Б-7-1963 20.11.67 | Минздрав РСФСР | |
| 71 | Ленты капроновые и хлопчатобумажные | В качестве стяжек пакетов для мясных туш и полутуш при механизированной перегрузке мяса | 123-9/39 07.03.64 | Минздрав СССР | ||
| 72 | Литопон и двуокись титана | В качестве красителя пластмасс при условии отсутствия растворимых солей бария и цинка, а также солей тяжелых металлов | 123-17/18 28.02.64 | Минздрав СССР | ||
| 73 | Мастика УМС-50 | В качестве прокладки под облицовочные детали холодильников | Полиизобутилен П-118, нейтральное масло и мел | 126-11/824-3 14.08.68 | Минздрав СССР | |
| 74 | Мешки джутовые | Для затаривания сахара | 123-9/585-7 24.05.65 | Минздрав СССР | ||
| 75 | Мешки сетчатые, пропитанные противогнилостными водостойкими составами | Для затаривания картофеля и др. овощей на время транспортировки | Латекс найрит Л-3, латексы: найрит Л-3+СКС30, латекс СКС30, сплав: парафин, канифоль, петролатум, антисептик — нафтенат меди | 08с/Б-7-965 02.04.60 | Минздрав РСФСР | |
| 76 | Мешки сетчатые, пропитанные противогнилостными водостойкими составами | Для хранения картофеля | Сплав ПКП, нафтенат меди, латекс найрит Л-3, латекс найрит Л-3+ СКС-30, латекс СКС-30 | 08с/Б-7-1283 15.07.60 | Минздрав РСФСР | |
| 77 | Мипора | ТУ МХП 2967-51 | В качестве изоляционного материала в холодильниках, предназначенных для хранения продуктов | 08с/Б-7-849 14.06.62 | Минздрав РСФСР | |
| 78 | Ойтисиковое масло, взамен ранее применявшегося тунгового масла | Применяется при изготовлении лака КР-1, предназначенного для покрытия внутренней поверхности банок под крабовые консервы | 08с/Б-7-1368 26.07.67 | Минздрав РСФСР | ||
| 79 | Оргстекло (полиметилметакрилат литьевой марки ЛПТ-1) | ТУ ТХП 63-47 | Доильные стаканы к доильному аппарату «Стимул» | Сополимер, включающий метилметакрилат и этилакрилат | 123-9/52 07.05.63 | Минздрав СССР |
| 80 | Пакеты бумажные, покрытые полиэтиленовой пленкой высокого давления марки П-2070-П | СТУ 36-10 -09-63 ГОСТ Ц0354-63 |
Для расфасовки сливок 10% и 20% жирности и сметаны 30% жирности | 08с/Б-7-366 03.03.64 | Минздрав РСФСР | |
| 81 | Паста уплотнительная (N 1 и N 2) | Для уплотнения пазов жестяных консервных банок | Рецепт пасты N 1: латекс квалитекс, сера, окись цинка, этилцимат, каолин, вазелин, редоксайд, карбоксиметилцеллюлоза, казеинат аммония, диспергатор НФ. Рецепт пасты N 2: латекс СКС-50К (СКС-50П), масло аммиачно-канифольное, ОП-10, сера, окись цинка, этилцимат, каолин, вазелин, редоксайд, карбоксиметилцеллюлоза, казеинат аммония, диспергатор НФ | 123-8/279-7 14.04.65 | Минздрав СССР | |
| 82 | Паста уплотнительная латексная с введением в нее эмульсии силиконового масла ПСМ-400 или импортной эмульсии в количестве 0,001% (с целью уничтожения вспенивания) | Для герметизации швов жестяной тары в консервной промышленности | -«- | 126-5/Б-37-3 18.06.69 | Минздрав СССР | |
| 83 | Пенопласт фенольно-резольный марки ФРП-1 | В качестве термоизоляционного слоя межобшивочного пространства кузова автофургона, предназначенного для перевозки пищевых продуктов | 126-14/24653 16.05.69 | Минздрав СССР | ||
| 84 | Пенополиуретан марки ППУ-305 <3> | В качестве теплоизоляции при изготовлении сомолетных холодильников СХШ-100 | Полиэфир (простой на основе ксилита и окиси пропилена), триэтиламин, добавка — блоксополимер на основе полидиметилсилоксана и окиси этилена, ФРЕОН-113 (1,1,2,-трифтортрихлорэтан), полиизоцианат на основе 4,4-дифенилметандиизоцианата | 126-14/20-3 12.06.68 | Минздрав СССР | |
| 85 | Пергамин, покрытый низкомолекулярным полиэтиленом высокого давления с двух сторон (мол. вес. 7000 — 8000, вес покрытия равен 25 г/м): покрытие наносится на бумажную поверхность путем пропускания бумажной ленты через расплав полиэтилена (120 град. C) на машине типа «Парафинера» | Для одноразовой упаковки только нежирных кондитерских изделий с влажностью не более 15% | 08с/Б-7б2508 21.06.68 | Минздрав РСФСР | ||
| 86 | Пластмасса — карболит К-18-2 | ГОСТ 5689-61 | Для изготовления опытной партии ручек для ножей | Фенолформальдегидная смола | 08с/Б-7-1244 18.07.67 | Минздрав РСФСР |
| 87 | Пластик полихлорвиниловый | Уплотнитель дверей домашних холодильников | Поливинилхлорид марки С-5, диоктилфталат очищенный, стеарат кальция, свинцовые белила, двуокись титана | 123-8/309-26 04.12.65 | Минздрав СССР | |
| 88 | Пластмасса «Декоррозит» | ВТУ М 796-58 | Для изготовления лопаток насоса для перекачки воды автомата «АТ-26», предназначенного для продажи газированной воды | Фенолформальдегидная смола, совмещенная с поливинилхлоридом, меламин, уротропин, каменноугольный кокс | 123-10/29 28.04.58 | Минздрав СССР |
| 89 | Пластобетон | Устройство опытных участков полов в плодоперерабатывающих предприятиях | На основе фурфуролацетонового мономера (Ф.А.) | 126-14/437-3 05.03.68 | Минздрав СССР | |
| 90 | Пленка поливинилхлоридная П-74 | Стаканчики для упаковки сметаны и творога | ПВХ-С60, полигард, CaO6, стеарат кальция, стеарат цинка, глицерин, диоктилфталат, эпоксидированное соевое масло, тинопал, двуокись титана | 126-14/564-3 19.08.69 | Минздрав СССР | |
| 91 | Пленка полиэтиленовая | ГОСТ 10354-63 | Для расфасовки свежемороженых плодов и овощей | 126-14/705-3 07.04.69 | Минздрав СССР | |
| 92 | Пленка полиэтиленовая | ГОСТ 10354-63 | 1. Для изготовления изделий для пищевой промышленности | 08с/Б-7-1069 22.05.64 | Минздрав РСФСР | |
| 2. Для обертки плавленых сыров | 123-17/106 31.03.54 | Минздрав СССР | ||||
| 3. Для упаковки сухого молока в трехслойные крафт-мешки с внутренним вкладышем из полиэтилена | 123-10/9 24.04.60 | Минздрав СССР | ||||
| 4. Для предохранения от усушки (хлеба, плодов) и для упаковки нежирных мясных и рыбных продуктов | 123-10/13 20.04.60 | Минздрав СССР | ||||
| 5. Для изготовления разовой упаковки под пищевые продукты | 113-17/35 05.11.52 | Минздрав СССР | ||||
| 6. Для изготовления вкладышей в сухотарные бочки под тузлучную рыбпродукцию | 08с/Б-9-1528 12.08.59 | Минздрав РСФСР | ||||
| 93 | Пленка полиэтиленовая (высокого давления) | ВТУ МХП 4430-50 | Для замораживания и хранения мяса | 123-9/20 28.02.64 | Минздрав СССР | |
| 94 | Пленка полиэтиленовая (полиэтилен марки П2115-11) | ГОСТ 10354-63 | Пакеты «тетрапак» под молочные продукты | 123-6/III-7 29.09.65 | Минздрав СССР | |
| 95 | Пленка полиэтиленовая нестабилизированная марки П2020Т | Для изготовления мешков под засолку овощей в бочках | 08с/Б-7б-864 05.06.69 | Минздрав РСФСР | ||
| 96 | Пленка ПЦ-2, ламинированная полиэтиленом бумага или кашированная фольгой | Для упаковки и расфасовки острокислых нестерилизуемых соусов | 08с/Б-7б2253 18.06.69 | Минздрав РСФСР | ||
| 97 | Пленка полимерная упаковочная на основе гидрохлорида синтетического каучука СКИ-3 («Эскаплен») | Упаковка пищевых продуктов | Гидрохлорид каучука, диоктилсебацинат, эпоксидированное соевое масло, сорбиновая кислота | 126-11/807/3 09.10.69 | Минздрав СССР | |
| 98 | Пленки на основе сополимера винилхлорида с винилиденхлоридом типа «Лаплен» разных рецептур (1, 2, 3, 4, 5, 6) <4> | Упаковка жидких и твердых пищевых продуктов, не содержащих жиров | 1. Сополимер винилиденхлорида с винилхлоридом высокой степени чистоты, дибутилсебацинат, ацетилтрибутилцитрат, карбоксилат, CaO6, стеарат магния | 126-5/415 29.08.69 | Минздрав СССР | |
| 2. Сополимер винилиденхлорида с винилхлоридом высокой степени чистоты, диоктилфталат, эпоксидированное соевое масло, бутилфталилбутилгликолят, Н-Г-22-46, стеарат магния | ||||||
| 3. Сополимер винилиденхлорида с винилхлоридом высокой степени чистоты, эпоксидированное соевое масло, бутилфталилбутилгликолят, CaO6, стеарин | ||||||
| 4. Сополимер винилиденхлорида с винилхлоридом высокой степени чистоты, эпоксидированное соевое масло, бутилфталилбутилгликолят, CaO6, стеарин, крахмал, диоктилфталат | ||||||
| 5. Сополимер винилиденхлорида с винилхлоридом высокой степени чистоты, эпоксидированное соевое масло, CaO6, стеарин, этилгексилакрилат | ||||||
| 6. Сополимер винилиденхлорида с винилхлоридом высокой степени чистоты, эпоксидированное соевое масло, CaO6, стеарин, полиэтилен |
| 99 | Пленка полиэтиленцеллофановая ПЦ-2 <5> | МРТУ 15-110-68 | Для упаковки рыбной кулинарии на Мосрыбкомбинате с соблюдением режимов и сроков реализации по ГОСТам, техническим условиям, инструкциям для следующих рыбпродуктов: | 126-14/10823 10.05.69 | Минздрав СССР | |
| 1. Балык из осетровых рыб | ||||||
| 2. Балык из дальневосточных лососевых х/к | ||||||
| 3. Лососи соленые внарезку | ||||||
| 4. Лососи дальневосточные соленые внарезку | ||||||
| 5. Рыба печеная | ||||||
| 6. Рыба жареная | ||||||
| 7. Котлеты рыбные жареные | ||||||
| 8. Сосиски рыбные и др. | ||||||
| 100 | Пленка целлофанполиэтилен марки П2070П | СТУ 102-141464 | 1. Вкладыш в фанерно-штамповочные бочки под сгущенное молоко с сахаром | N 613 23.09.65 | Минздрав УССР | |
| 2. Для упаковки бескоркового сыра при прямом контакте с сыром | 08с/Б-7-1510 12.07.63 | Минздрав РСФСР | ||||
| 101 | Пленка полимерная дублированная — полиэтилен, целлофан ПЦ-1, ПЦ-2 | МРТУ 18/180-67 (МЗ СССР 24.03.67) | Для упаковки открытой карамели и драже | 08с/Б-7б-335 04.02.68 | Минздрав РСФСР | |
| 102 | Пленка целлофанполиэтилен марки П2070П и П2115П нестабилизированный | СТУ 1021414-64 | Для упаковки хлеба на длительное хранение | 123-11/26614 01.03.65 | Минздрав СССР | |
| 103 | Подпергамент, бумага, ламинированная полиэтиленом марки П2070П | МРТУ 6-05-88965 | Упаковка чая | 08с/Б-7б-293 06.03.69 | Минздрав РСФСР | |
| 104 | Подпергамент, покрытый низкомолекулярным полиэтиленом высокого давления с двух сторон (мол. вес 7000 — 8000), вес покрытия равен 25 г/м; покрытие наносится на бумажную поверхность путем пропускания бумажной ленты через расплав полиэтилена (до 120 град.) на машине типа «Парафинера» | ГОСТ 1760-58 | Для упаковки кондитерских изделий | 08с/Б-7б2508 21.06.68 | Минздрав РСФСР | |
| 105 | Покрытие из перхлорвиниловой смолы без наполнителя и с наполнителем — двуокисью титана <6> | ГОСТ 10004-62 | Защита винных емкостей от действия агрессивной среды | Смола перхлорвиниловая | 126-14/400-3 21.02.69 | Минздрав СССР |
| 106 | Покрытие эмалевое с предварительной шпаклевкой | Для внутренней поверхности бочек под тузлучные рыбопродукты | Состав эмали: парафин очищенный, канифоль ГОСТ 797-64 Состав шпаклевки: казеин, гашеная известь, жидкое стекло, алебастр | 113-17/106 14.07.51 | Минздрав СССР | |
| 107 | Покрытие 3-слойное на основе эпоксидной смолы ЭД-5 и ЭД-6 | Для окраски внутренней поверхности экстрактора, используемого в масложировой промышленности | 1. Слой — грунт ЭП-01 красный: эпоксидная смола ЭД-5, сурик железный, растворитель Р-4 (толуол, бутилацетат, ацетон) | 126-11/128-3 27.03.69 | Минздрав СССР | |
| 2. Слой — шпаклевка ЭП-00-2; смола эпоксидная ЭД-5, каолин | ||||||
| 3. Слой — эмаль ЭМ-73 белая: смола эпоксидная ЭД-6, двуокись титана (малярная, пигментная), растворитель Р-4 (толуол, бутилацетат, ацетон) | ||||||
| 108 | Покрытие на основе смолы ЭД-5 <7> | ГОСТ 10587-63 | Для защиты внутренней поверхности дошников для хранения капусты | Дифенилолпропан, эпихлоргидрин, полиэтиленполиамин, ацетон | 08с/Б-7-354 29.07.69 | Минздрав РСФСР |
| 109 | Покрытие на основе эпоксидных смол | ГОСТ 10587-63 | Для покрытия емкостей, предназначенных для хранения соков, безалкогольных напитков, а также и для применения в молочной промышленности | 1 слой — грунт ЭС: эпоксидная смола ЭД-5, железный сурик, ацетон, полиэтиленполиамин | 123-15/22 22.03.62 | Минздрав СССР |
| 2 слой — грунт ЭК (наносится на грунт ЭС): эпоксидная смола ЭД-5, каолин, полиэтиленполиамин | ||||||
| 3 слой — эмаль ЭТК (наносится на грунт ЭК): эпоксидная смола ЭД-6, двуокись титана, ацетон, полиэтиленполиамин | ||||||
| 110 | Покрытие эпоксидное | Для заделки швов внутренней обшивки рефрижераторных камер (в условиях опытной эксплуатации) | 1 слой (промазочный) — эпоксидный компаунд К-153, эпоксидная смола ЭД-5, жидкий тиокол, полиэфиракрилат, полиэтиленполиамин | 08с/Б-7б-889 11.03.68 | Минздрав РСФСР | |
| 2 слой (основной) — эпоксидный компаунд К-153, алюминиевая пудра, полиэтиленполиамин, армирующий материал — стеклоткань бесцветная марка АСТ-Т(б) | ||||||
| 3 слой (отделочный) — эпоксидный компаунд К-153, алюминиевая пудра, полиэтиленполиамин | ||||||
| 111 | Полиакриламид (с содержанием свободного мономера не выше 0,5%) | 1. Для осветления вин | 126-14/24143 12.02.69 | Минздрав СССР | ||
| 2. Применяется для пропитки мешков при их изготовлении | 123-14/343-7 26.05.65 | Минздрав СССР | ||||
| 3. Коагулянты для очистки соков первой сатурации в свеклосахарном производстве | 126-11/359-3 15.03.68 | Минздрав СССР | ||||
| 112 | Полиакриламид (с содержанием мономера не выше 0,5% в комплексе с бетонитом) <8> | Интенсификация осветления и стабилизации виноградных и плодоягодных вин | 126-14/174-3 07.03.68 | Минздрав СССР | ||
| 113 | Поливинилхлоридные трубки | В качестве покрытия крюков троллей для навешивания мясных туш | Поливинилхлорид, смола поливинилхлоридная, дибутилфталат, окись цинка, стеарат кальция | 06с-3А/1152 06.06.64 | Минздрав Молдавской ССР | |
| 114 | Поливинилхлорид марки Ш-46 | Шланги доильных агрегатов. Кратковременный (минуты) контакт с молоком | Поливинилхлорид, диоктилфталат, эпоксидированное соевое масло, стеарат кальция, стеарат цинка, топанол | 126-11/698-3 27.08.69 | Минздрав СССР | |
| 115 | Поливинилхлорид марки Ш-62 | То же | Поливинилхлорид, диоктилфталат, эпоксидированное соевое масло, стеарат кальция, стеарат цинка, топанол, пигмент голубой фталоцианиновый | 126-11/698-3 27.08.69 | Минздрав СССР | |
| 116 | Полиизобутилен П-20 | Уплотнительные прокладки к кроненпробкам пиво-безалкогольных напитков | Полиизобутилен П-20, каолин, литапон | 3/7920 23.08.63 | Минздрав Латвийской ССР | |
| 117 | Полиметилметакрилат с добавлением 2 и 4% бутилакрилата | Трубы доильных аппаратов | Чистый полиметилметакрилат, сополимер метилметакрилата с бутилакрилатом | 126-11/698-3 27.08.69 | Минздрав СССР | |
| 118 | Полиметилметакрилат литьевой ЛПТ-1 | МРТУ 6М 871-62 | Для изготовления деталей кухонной и посудомоечной машин | 126-14/20423 19.09.68 | Минздрав СССР | |
| 119 | Полиметилметакрилат марки ЛСОМ | ВТУ П-126-67 | Для соковыжималки — корпус, толкатель, крышка и стакан для сбора сока; для миксера — стакан; для овощерезки — корпус, толкатель и крышка; для посудомоечной машины материал может быть использован, если заказчика не смущает появление матовости материала при контакте с горячей водой | Метилметакрилат, додецил- или лауирилмеркаптан. Порофор (азодиизобутилонитрил) | 126-11/174-3 01.09.68 | Минздрав СССР |
| 120 | Полимерцементные составы на найрите Л-ЗП и на латексе ДВХБ-70 | Для крепления керамических плиток на рыбопромысловых судах | 126-14/25203 12.02.69 | Минздрав СССР | ||
| 121 | Полипропилен марки ПП-1 <9> | СТУ 3613-126-65 | Для соковыжималки и овощерезки — толкатель и крышки; для посудомоечной машины — корпус и крышка | CaO6, тинувин, двуокись титана | 126-11/174-3 01.09.69 | Минздрав СССР |
| 122 | То же | СТУ 3613-126-65 | Для изготовления деталей кухонной и посудомоечной машин | 126-14/20423 19.09.68 | Минздрав СССР | |
| 123 | Полипропилен марки Л6П10/040. Стабилизированный CaO6 (бис-5,метил-3третбутил-2, оксифенилмоносульфид) при содержании атактической фракции до 1,0%, индекс расплава 3,5 — 4,5 г/10 мин. | МРТУ 6-05-1105 -67 | Изготовление ткани для фильтрования творога от сыворотки при условии обработки ткани перед эксплуатацией 1% раствором кальцинированной соды (в течение 30 мин.), затем горячей водой до полного удаления моющего раствора и стерилизации кипячением в течение 10 — 15 мин. | 126-14/11653 29.10.69 | Минздрав СССР |
| 124 | Полипропилен марки 04П01, стабилизированный дилаурилтиодипропионатом и топанолом СА при содержании атактической фракции до 2% | МРТУ 605-110567 | Изготовление деталей молочного сепаратора | 126-14/19653 31.10.69 | Минздрав СССР | |
| 125 | Полипропилен марок 01П10/002; 02П10/003 при индексе расплава 0,2 — 0,3 г/10 мин. с содержанием атактической фракции до 2% | МРТУ 605-110567 | Крышки для домашнего консервирования | 08с/Б-7б1414 12.07.68 | Минздрав РСФСР | |
| Для изготовления изделий, соприкасающихся с пищевыми продуктами | 08с/Б-7б-293 29.03.68 | Минздрав РСФСР | ||||
| 126 | Полипропилен | Для изготовления подносов в общественном питании | 08с/Б-7б2078 09.10.67 | Минздрав РСФСР | ||
| 127 | Полистирол блочный марки «Д», прозрачный неокрашенный | ГОСТ 9440-60 | 1. Для изготовления изделий «Взбивалка» | Стирол | 126-14/21463 21.10.68 | Минздрав СССР |
| 2. Крышки к ручной картофелечистке | 08с/Б-7-416 25.02.64 | Минздрав РСФСР | ||||
| 128 | Полистирол блочный марки «Т» | Для изготовления пельменниц | 08с/Б-7б-535 20.03.67 | Минздрав РСФСР | ||
| 129 | Полистирол блочный и суспензионный | ГОСТ 9440-60 МРТУ 6-05-92864 |
Конфетницы, сахарницы, подносы, хлебницы и т.д. | 08с/Б-7-437 18.03.64 | Минздрав РСФСР | |
| 130 | Полистирол блочный и эмульсионный | ГОСТ 9440-60 | 1. Сахарницы, подносы, конфетницы, хлебницы | 123-9/12 03.03.64 | Минздрав СССР | |
| 2. Для изготовления ручек и футляров к зубным щеткам | 123-9/57 25.03.64 | Минздрав СССР | ||||
| 3. Для внутренней облицовки домашних холодильников | 08с/Б-2-538 23.03.64 | Минздрав РСФСР | ||||
| 131 | Полистирол компаундированный марки «ПК» (производство Кусковского химзавода) | ВТУ Г-103 -67 | 1. Изготовление блюдец-подносов (изделия N 5282) | Полистирол суспензионный, каучук, масло вазелиновое или бутилстеарат, двуокись титана | 126-14/56-3 08.01.68 | Минздрав СССР |
| 2. Для изготовления изделий «Взбивалка» | 126-14/21463 21.10.68 | Минздрав СССР | ||||
| 132 | Полистирол ударопрочный суспензионный марки ПС-СУ (на основе каучука «Интен») | Изготовление деталей домашнего холодильника | 126-5/15-3 10.01.68 | Минздрав СССР | ||
| 133 | Полистирол ударопрочный суспензионный марки ПС-СУ2 (содержание остаточного мономера — стирола не более 0,1%) | МРТУ 605-956-69 | 1. Терки для фруктов и овощей | Стирол, каучук («Интен» 55 или СКД ЛП) (пластификатор — масло вазелиновое медицинское, краситель — двуокись титана, стабилизатор — фосфит НФ, стеарат цинка) | 08с/Б-7-Б945 16.08.67 | Минздрав РСФСР |
| 2. Для изготовления изделий «Взбивалка» 3. Для внутренней облицовки домашних холодильников | 126-14/21463 21.10.68 123-9/3 15.01.64 08с/Б-2-538 23.03.64 |
Минздрав СССР Минздрав СССР Минздрав РСФСР | ||||
| 4. Для производства изделий широкого потребления | 123-9/12 03.03.64 | Минздрав СССР | ||||
| 5. Противни конвейера, по которому движется салака | 3/3-204 07.06.64 | Минздрав Латвийской ССР | ||||
| 6. Лотки и подносы | 08с/Б-7-561 23.03.64 | Минздрав РСФСР | ||||
| 134 | Полистирол марки СНП | Лотки для установления банок со специями, ящики для кастрюль, скобы для крышек, лотки для сервирования столов, лотки для сыпучих продуктов, бункер для овощей | 08с/Б-7-2293 03.10.64 | Минздрав РСФСР | ||
| 135 | Полистирол марки СНП-2 (содержание остаточных мономеров: стирола не более 0,5%, акрилонитрила не более 0,01%) | СТУ 77-494-62 СТУ 3613-858-62 |
Для внутренней облицовки холодильника | Сополимер стирола с акрилонитрилом, каучук СКН-26, пластификатор — дибутилфталат, стабилизатор — алкофен Б, краситель — двуокись титана, смазка — стеарат кальция | 126-Н/8-1033 27.08.64 | Минздрав СССР |
| 136 | Полистирол марки СНП-2П (содержание остаточных мономеров: стирола не более 0,25%, акрилонитрила не более 0,008%) | 1. Для изготовления головок автосифонов (контакт с водой кратковременный) | Сополимер стирола с акрилонитрилом, каучук СКН-26 (бутадиенстирольный) | 126-14/24003 25.12.68 | Минздрав СССР | |
| 2. Упаковка пищевых продуктов, имеющих влажность не более 55%, со сроком хранения не более 10 дней (сыры типа «Янтарь», «Сливочный», творог, мясо, копченая, сырая и присоленная рыба, сухие и сыпучие продукты) | 2,4,6-три-третбутилфенол, масло вазелиновое медицинское или дибутилсебацинат, стеарат кальция | 126-14/12343 11.08.69 | Минздрав СССР | |||
| 137 | Полистирол марки СНП-К (пластифицированный диоктилфталатом) | Для упаковки сыпучих пищевых продуктов, свежих и сухих фруктов, ягод и овощей, детских игрушек, предметов широкого потребления (за исключением тарелок, бытовых емкостей, фляг и пр., игрушек для детей ясельного возраста) | 126-5/478-3 02.01.69 | Минздрав СССР | ||
| 138 | Полистирол ударопрочный марки УП-1 | 1. Лотки и подносы | 08с/Б-7-561 23.03.64 | Минздрав РСФСР | ||
| 2. Для внутренней облицовки холодильников | 126-Н/8-1033 27.08.64 | Минздрав СССР | ||||
| 139 | Полистирол ударопрочный марки УП-1Л <10> | МРТУ 605-956-67 | 1. Для изготовления деталей электромиксера «Метеор» | 08с/Б-7б1636 30.09.67 | Минздрав РСФСР | |
| 2. Для изготовления изделий «Взбивалка» | 126-14/21463 21.10.68 | Минздрав СССР | ||||
| 3. Изготовление шинковки | 126-14/538-3 17.05.68 | Минздрав СССР | ||||
| 140 | Полистирол ударопрочный марки УП-1Э (содержание остаточного мономера стирола не более 0,55%) | МРТУ 605-956-69 | 1. Изготовление внутреннего шкафа, панели дверей и дверец холодильной камеры домашних холодильников | Стирол, каучук, СКД, стеарат цинка, перекись дитретичного бутила, гидроперекись кумола; перекись дикумила, нормальный лаурилмеркаптан, вазелиновое масло, двуокись титана, голубой фталоцианиновый, полигард, стеариновая кислота | 123-5/151 02.03.68 | Минздрав СССР |
| 2. Емкости для холодильника «Тамбов» | 123-5/91-3 20.08.69 | Минздрав СССР | ||||
| 141 | Полистирол ударопрочный марки УПП (содержание остаточного мономера стирола не более 0,5%) | Для внутренней облицовки холодильников | Стирол, каучук, «Интен» 55 N «А» или СКД-ЛП, пластификатор — масло вазелиновое медицинское, стеариновая кислота, стеарат цинка; стабилизаторы — беназол П и фосфит НФ | 126-Н/8-1033 27.08.64 | Минздрав СССР | |
| 142 | Полистирол ударопрочный на каучуке СКД марки УПП-1СТ (содержание остаточного мономера стирола не более 0,37%) | Емкости для холодильника «Тамбов» | Стирол, каучук СКД, стеариновая кислота, стеарат цинка, Н-лаурилмеркаптан, вазелиновое масло, перекись дитретичного бутила, двуокись титана, голубой фталоцианиновый, полигард | 123-5/91-3 20.08.68 | Минздрав СССР | |
| 143 | Полистирол ударопрочный марки УПП-2СТ <11> | СТУ 3613-153-65 | 1. Для использования в кондитерской промышленности | 123-5/125-3 20.03.69 | Минздрав СССР | |
| 2. Подносы под чашки (контакта с пищевыми продуктами не имеют) | 126-14/18273 03.01.69 | Минздрав СССР | ||||
| 144 | Полиэтилен высокого давления (низкой плотности) марок 111ПЭ20/020Т, 112ПЭ20/020П | Для изготовления тары под рыбопродукцию | 126-5/583-3 30.12.68 | Минздрав СССР | ||
| 145 | Полиэтилен высоко | МРТУ 6-05-88962 | Изготовление крышек к стеклянным банкам | 08с/Б-71227/866 25.05.64 | Минздрав РСФСР | |
| кой плотности) нестабилизированный марки П2035Т | ||||||
| 146 | Полиэтилен высокого давления (низкой плотности) марок 112ПЭ20/020П, 147ПЭ20/020В, полученный с помощью перекисных катализаторов | В контакте с пищевыми продуктами влажностью не выше 15%, с питьевой холодной водой (кратковременно) | 126-14/ 215713 19.07.68 | Минздрав СССР | ||
| 147 | Полиэтилен высокого давления (низкой плотности) марок 112ПЭ20/020Н, 147ПЭ20/020В | Изготовление пленок, предназначенных для контакта с пищевыми продуктами | 126-14/ 215713 19.07.68 | Минздрав СССР | ||
| 148 | Полиэтиленовые вкладыши (полиэтилен высокого давления — низкой плотности) марок П2020П и П2070П | Для жестяных банок N 14 и 15 емкостью 3 — 8 л, покрытых лаком ЭП-527, под расфасовку томатпасты и повидла | 126-14/10473 03.06.68 | Минздрав СССР | ||
| 149 | Полиэтилен высокого давления (низкой плотности) марок 1085-67, 111ПЭ20/020Т, 113ПЭ20/020 и 142ПЭ20/020П | Емкости без крышек для контакта с пищевыми продуктами с влажностью до 15% | 126-5/374-3 07.08.68 | Минздрав СССР | ||
| 150 | Полиэтилен высокого давления (низкой плотности) марки П2070П | МРТУ 6-05-88965 | Изготовление тазиков для заморозки мясных блоков при 18 — 23 град. C в течение 24 — 48 час. | 126-14/387-3 22.02.68 | Минздрав СССР | |
| 151 | Полиэтилен высокого давления (низкой плотности) стабилизированный | Для изготовления опытного подземного молокопровода на участке от фермы к экспериментальному заводу ВНИИ (г. Углич) | Стабилизирован CaO6: бис-(5-метил-3-третбутил- 2-оксифенил) — моносульфид или 2246: бис-(5-метил-3-третбутил- 2-оксифенил) метан (бисалкофен БП) |
126-14/25993 1968 | Минздрав СССР | |
| 152 | Полиэтилен высокого давления (низкой плотности) нестабилизированный марки П2010В | Тазики-лотки для транспортировки и реализации студня | 126-14/557-3 19.04.68 | Минздрав СССР | ||
| 153 | Полиэтилен высокого давления (низкой плотности) нестабилизированный марки П2020Т | МРТУ 6-05-88965 | 1. Ванночки для пищевого льда в домашних холодильниках | 08с/Б-7-2645 21.03.66 | Минздрав РСФСР | |
| 2. Для изготовления изделий «Взбивалка» | 126-14/21463 21.10.68 | Минздрав СССР | ||||
| 3. Кружки, лейки (с обязательным оттиском «для холодной воды») | 08с/Б-7б1027 25.07.68 | Минздрав РСФСР | ||||
| 4. Тазики для заморозки блоков мяса | 08с/Б-7б-502 12.04.68 | Минздрав РСФСР |
| 154 | Полиэтилен высокого давления (низкой плотности) марки П2035 | МРТУ 6-05-88965 | Для изготовления изделий «Взбивалка» | 126-14/21463 21.10.68 | Минздрав СССР | |
| 155 | Полиэтилен высокого давления (низкой плотности) марки П2020Т | МРТУ 6-05-88965 | 1. Изготовление крышек к стеклянным банкам | 08с/Б-71227/866 25.05.64 | Минздрав РСФСР | |
| 2. Для изготовления крышек к стеклянным банкам с жидкой горчицей | 08с/Б-7б-71 17.01.68 | Минздрав РСФСР | ||||
| 156 | Полиэтилен высокого давления (низкой плотности) марок П2020Т, П2018В | МРТУ 6-05-88965 | 1. Для изготовления пакетов для разлива молока | Пигментированный двуокисью титана (марка РО-2 ГОСТ 9808-65) и содержащий скользящую добавку (амид олеиновой кислоты) | 126-14/18253 29.11.68 | Минздрав СССР |
| 2. Упаковка творога <12> | Пигментированный двуокисью титана (марка РО-2 ГОСТ 9808-65) и содержащий скользящую добавку (амид олеиновой кислоты) | 126-14/22033 23.10.69 | Минздрав СССР | |||
| 157 | Полиэтилен высокого давления (низкой плотности) марок П2020Т и П2035Т | МРТУ 6-05-88965 | Крышки | 08с/Б-7-1933 15.08.64 | Минздрав РСФСР | |
| 158 | Полиэтилен высокого давления (низкой плотности) П2020Т, защищенный пленкой целлофанполиэтилен | МРТУ 6-05-88965 СТУ 102-141464 | Для хранения в течение 4 месяцев яблочного сока в пакетах, расфасованного в асептических условиях | N 689 09.02.65 | Минздрав УССР | |
| 159 | Полиэтилен высокого давления (низкой плотности) марки П2070П | МРТУ 6-05-88965 | 1. Для покрытия бумажных пакетов «тетрапак» под кефир | 08с/Б-7-1476 09.07.63 | Минздрав РСФСР | |
| 2. Для покрытия бумажных пакетов «тетрапак» под молоко | 08с/Б-7-24 07.01.63 | Минздрав РСФСР | ||||
| 160 | Полиэтилен высокого давления (низкой плотности) нестабилизированный марки П2070П (индекс расплава 5,4 г/10 мин.) | МРТУ 6-05-88965 | Фляги, полученные методом спекания полиэтиленового порошка при температуре 160 — 190 град. C в среде азота | 123-9/145 30.09.64 | Минздрав СССР | |
| 161 | Полиэтилен высокого давления (низкой плотности) марки П2020Т | ГОСТ 10354-63 МРТУ 6-05-88965 | Вкладыши в сухотарные бочки при упаковке, перевозке и хранении сельди и соленой рыбы | 123-9/40 27.04.63 | Минздрав СССР | |
| 162 | Полиэтилен высокого давления (низкой плотности) нестабилизированный и неокрашенный марок: П2020Т, П2010В, П2035Т, П2006 | МРТУ 6-06-88965 | Крышки в условиях домашнего консервирования (только для укупорки варенья, джема, повидла, солений — огурцов и помидоров, а также компотов для недлительного хранения <13> | 126-5/217-3 28.07.69 | Минздрав СССР | |
| 163 | Полиэтилен нестабилизированный высокого давления (низкой плотности) марок П2020Т, П2010В, П2006 | Для хранения морковки | 126-14/13293 24.06.69 | Минздрав СССР | ||
| 164 | Полиэтилен высокого давления (низкой плотности) марок: 111ПЭ20/020Т 113ПЭ20/020О 115ПЭ20/070В 128ПЭ20/040П 133ПЭ20/003Л 148ПЭ20/007В |
МРТУ 6-051085-67 | Емкости без крышек для контакта с пищевыми продуктами с влажностью до 15% <14> | Полученных с применением перекисных катализаторов (дитретбутил перекиси лаурила 0,017% и третбутилпербензоата — 0,03%) | 126-11/731а3 22.09.69 | Минздрав СССР |
| 165 | Полиэтилен высокого давления (низкой плотности) марки П2010В нестабилизированный | МРТУ 6-05-88965 | Для изготовления пакетов и пленок, предназначенных для упаковки и транспортировки сахара в страны с тропическим климатом | 126-14/577-3 10.03.69 | Минздрав СССР | |
| 166 | Полиэтилен высокого давления (низкой плотности) неокрашенный | МРТУ 6-05-88965 | Для изготовления фляг емкостью 1 л при условии выштамповки «для холодной воды» | 08с/Б-7б-845 16.06.69 | Минздрав РСФСР | |
| 167 | Полиэтилен высокого давления (низкой плотности) марки П2020Т | 1. Термосы с изоляционным кожухом к самоходным комбайнам СК-4 | 08с/Б-7б-301 04.03.69 | Минздрав РСФСР | ||
| 2. Вкладыши в бочки для квашения капусты | 08с/Б-7б1290 09.09.69 | Минздрав РСФСР | ||||
| 3. Тары для транспортировки творога, сырков (расфасованных) | 08с/Б-7б1440 28.03.69 | Минздрав РСФСР | ||||
| 168 | Полиэтилен высокого давления (низкой плотности) марки 112ПЭ-20/020П | Тубы под горчицу | 08с/Б-7б1451 14.08.69 | Минздрав РСФСР | ||
| 169 | Полиэтилен низкого давления (высокой плотности) марки П4020Э | МРТУ 6-05-89065 | Ручки к зубным щеткам | 10/4-443 31.12.64 | Минздрав Латвийской ССР | |
| 170 | Полиэтилен низкого давления (высокой плотности) марки П4040П | МРТУ 6-05-89065 | Секционные рамки для нужд пчеловодства | 08с/Б-7-174/ 1428 17.06.64 | Минздрав РСФСР | |
| 171 | Полиэтилен низкого давления (высокой плотности) П4007Э (индекс расплава в пределах 0,7 — 0,9) и полипропилен марок ПП-4, ПП-5 (индекс расплава в пределах 1,5 — 2,0) с содержанием атактической фракции до 2%, нестабилизированный | Изготовление лотков для перевозки фруктов и овощей авиатранспортом | 126-14/22613 24.01.68 | Минздрав СССР | ||
| 172 | Полиэтилен низкого давления (высокой плотности), стабилизированный стеаратом кальция | МРТУ 6-05-89065 | Изготовление лотков для транспортировки и хранения хлебобулочных изделий | 126-14/750-3 05.04.68 | Минздрав СССР | |
| 173 | Полиэтилен низкого давления (высокой плотности) марки П4007 | МРТУ 6-05-89065 | Рекомендованы для изготовления крышек для домашнего консервирования | 08с/Б-7б1489 10.08.67 | Минздрав РСФСР | |
| 174 | Полиэтилен низкого давления (высокой плотности) марки 20506-007 (индекс расплава в пределах 0,4 — 0,9) нестабилизированный и стабилизированный CaO6 | МРТУ 6-05-89067 | Для изготовления изделий, соприкасающихся с пищевыми продуктами | 08с/Б-7б-293 29.03.68 | Минздрав РСФСР | |
| 175 | Полиэтилен низкого давления (высокой плотности) | МРТУ 6-05-89064 | Для изготовления подносов для предприятий общественного питания, совков для сыпучих товаров, ящиков для перевозки расфасованного и завернутого мороженого и полуфабрикатов | 08с/Б-7-2662 22.10.64 | Минздрав РСФСР | |
| 176 | Полиэтилен-лавсан | Для контакта с пищевыми влажными продуктами при температуре 50 — 60 град. C, а также для упаковки пищевых концентратов, содержащих не более 15% жира | 126-14/624-3 27.06.69 | Минздрав СССР | ||
| 177 | Полиэтилен-целлофан | Для контакта с пищевыми влажными продуктами при температуре 40 град. C, а также для упаковки пищевых концентратов, содержащих не более 15% жира | 126-14/624-3 27.06.69 | Минздрав СССР | ||
| 178 | Полиэтиленовая пленка нестабилизированная марок «А» и «Б» | Пакеты для расфасовки и упаковки макаронных изделий | 08с/Б-7-1623 05.09.66 | Минздрав РСФСР | ||
| 179 | Полиэтиленовая пленка (толщиной 200 микрон) | Для изготовления мешков, предназначенных для упаковки соли на экспорт | 126-14/1449 03.07.68 | Минздрав СССР | ||
| 180 | Прокладка в кроненпробки и в алюминиевые колпачки | Для укупорки ликеро-водочных и безалкогольных напитков | Полиизобутилен П-200 (ГОСТ 13303-67), полиэтилен высокого давления марки П2070П, бумага СТУ 30-63-32-69, поликремневая кислота (белая сажа) марки У-333, парафин марки П-1 (ГОСТ 13577-68) | 08с/Б-7-1190 1969 | Минздрав РСФСР | |
| 181 | Смолы ионообменные КУ-2 и АВ-16Г | Для очистки раствора при производстве пищевой лимонной кислоты | 126-5/205-3 27.07.68 | Минздрав СССР | ||
| 182 | Смола обесфеноленная фенолформальдегидная | Для покрытия и склеивания фанеры, идущей на изготовление бочек, предназначенных под пищевые продукты | Состав: фенол, формалин, едкий натр, вода | 113-17/173 28.07.53 | Минздрав СССР | |
| 183 | Смола эпоксидная марки ЭД-6, отвержденная полиэтиленполиамином | Для покрытия внутренних поверхностей, стен и пола складов бестарного хранения сахарапеска | 126-14/138-3 02.04.69 | Минздрав СССР | ||
| 184 | Смола эпоксидная | Для покрытия внутренних поверхностей железобетонных сборных элементов мучных силосов | ЭД-5, кварцевый песок, мармалит, полиэтиленполиамин | 126-14/681-3 18.12.68 | Минздрав СССР | |
| 185 | Смола «Резит» | Для изготовления мундштуков | Фенолформальдегидная смола | 123-10/17 19.11.59 | Минздрав СССР | |
| 186 | Смола 101 | ТУ МХП КУ-328-53 | 1. Для наклейки этикеток на бутылки с пивом и безалкогольными напитками | Паратретичный бутилфенол, формальдегид | 3/3-106 19.02.64 | Минздрав Латвийской ССР |
| 2. Для наклейки этикеток на оригинальную упаковку в молочно-консервном производстве | 3/3-106 19.02.64 | Минздрав Латвийской ССР | ||||
| 187 | Смолка Л-3 | ТУ МХП 240-60 | Для наружного опечатывания пробок посуды с винно-водочными изделиями | 08с/Б-7-614 14.05.62 | Минздрав РСФСР | |
| 188 | Сополимер МСН-1 | ГОСТ 12271-66 | Для соковыжималок — толкатель крышки; для овощерезки — толкатель крышки; для посудомоечной машины — крышки | Метилметакрилат, стирол, нитрил акриловой кислоты, перекись бензоила, додецилили лаурилмеркаптан | 126-11/174-3 01.09.69 | Минздрав РСФСР |
| 189 | Спирт этиловый технический (гидролизный) | ГОСТ 8314-57 | 1. Для растворения анилиновых красителей, наносимых на пищевую тару (стаканчики бумажные, коробочки для мороженого, соли и т.д.) | 08с/Б-7-110 21.01.64 | Минздрав РСФСР | |
| 2. Для растворения красителей, применяемых для окраски курительных трубок из древовидного корня вереска, бриар и буковых, а также для окраски и промывки деталей мундштуков из органического стекла | 123-14/201 07.02.64 | Минздрав СССР | ||||
| 3. Для растворения красителей при окрашивании алюминиевой фольги | 123-9/24 20.02.64 | Минздрав СССР |
| 190 | Стабилизаторы: 2-окси-4-алк (С7-9) оксибензофенон (бензон ОА), 2-окси-4-октоскибензофенон (бензон ОО), бис-(5-метил-3-третбутил- 2-оксифенил)-метан (бисалкофен БП), 2,6-диизоборнил- 4-метилфенол (алкофен ДИП) | ВТУ 16-64 СТУ 36-13-3264 | В качестве стабилизаторов пластмасс, используемых в пищевой промышленности, водоснабжении, для изготовления детских игрушек | Вводится в количествах от 0,01% до 1,0% | 123-9/135 18.11.64 | Минздрав СССР |
| 190 а | Стабилизатор CaO6, бис-(5-метил-3-третбутил -2-оксифенил)-моносульфид (тиоалкофен БП) | ВТУ 16-64 | При производстве синтетических материалов, контактирующих с пищевыми продуктам | 123-9/132 15.08.64 | Минздрав СССР | |
| 191 | Бумажные стаканчики (покрытые парафином марки А и Б) | ГОСТ 784-53 | Упаковка зерненого творога (жирность 6%) | 123-14/794-7 15.05.67 | Минздрав СССР | |
| 192 | Стекло жидкое | ГОСТ 962-41 | Для силикатирования бочек под топленое масло | Силикат натрия | 113-14/65 30.01.52 | Минздрав СССР |
| 193 | Стекло органическое марки ПА с остаточным количеством мономера в готовом изделии 0,2 — 0,3% и при условии отмывки мономера — метилового эфира метакриловой кислоты в течение 6 суток проточной водой с температурой 40 град. C | ТУ 26-54 | 1. Емкости для домашних холодильников 2. Для изготовления молокопроводов на молокозаводах и деталей, соприкасающихся с молоком | Метиловый эфир метакриловой кислоты, перекись бензоила, дибутилфталат, крупка, стружка ММА | 08с/Б-7б-985 24.06.69 123-7/34 17.04.61 | Минздрав РСФСР Минздрав СССР |
| 194 | Стекло органическое | Для ящиков в качестве внутрицеховой тары под ястыки кетовой икры | 123-10/55 15.07.58 | Минздрав СССР | ||
| 195 | Стеклопластик | В качестве покрытия крюков троллей для навешивания мясных туш | Стекловолокно — эпоксиднодиеновая смола ЭД-6, ацетон, полиэтиленполиамин, дибутилфталат | 06с-3А/1152 06.06.64 | Минздрав Молдавской ССР | |
| 196 | Стеклопластик | В конструкциях аспирационных камер зерноочистительных машин (необходимо, чтобы отходы не задерживались в камере более 3 суток и не использовались в составе комбикорма для скота) | Стеклоткань АСИИ (сигма)-С2 (МРТУ 6М-814), стекложгут (МРТУ 6-05396-63) Полиэфирная смола ПН-1 (ВТУ ЛСНХ 33085-60) Нафтенат кобальта (ВТУ ЛСНХ 3110460); двуокись титана (ВТУ ЛСНХ 30022-59); гипериид (ТУ N БУ-11-53); шпаклевка ЦН008 (ТУ МХП 958-47) |
126-14/2457 07.05.69 | Минздрав СССР | |
| 197 | Сульфанол | Для мытья посуды на предприятиях общественного питания | 123-7/57 24.05.61 | Минздрав СССР | ||
| 198 | Сурик железный на олифе | ГОСТ 8135-62 ГОСТ 7931-56 |
Для покрытия цистерн и водонапорных баков | 113-17/125 30.03.54 | Минздрав СССР | |
| 199 | Сурик свинцовый на олифе | ГОСТ 1787-50 | Для покрытия ребристых охлаждающих частей холодильников | 113-17/59 18.11.54 | Минздрав СССР | |
| 200 | Титан двуокись | ВТУ 410-54 | В качестве красителя пластмасс при условии отсутствия растворимых солей бария и цинка, а также солей тяжелых металлов | 123-17/18 28.09.64 | Минздрав СССР | |
| 201 | Ткань капроновая арт. 1559 с пропиткой кремнийорганическим каучуком СКТ | Для фильтрации соков и сиропов при условии: содержание в капроновом волокне остаточного мономера капролактама не должно превышать 1%; капроновая ткань после пропитки СКТ должна быть промыта в течение 2 часов при температуре 30 град. для удаления примесей | 123-5/231-3 27.08.68 | Минздрав СССР | ||
| 202 | Фанера марки ФСФ и ФК, склеенная мочевиноформальдегидной смолой марки М-19-62 | МРТУ 13-06-467 | Шинковки для капусты | 08с/Б-7-1204 17.07.68 | Минздрав РСФСР | |
| 203 | Фенопласт К-21-22 (фенолит) | ГОСТ 5689-60 | Корпуса электродвигателей к универсальной бытовой машине «Белка» | 08с/Б-7-2397 11.10.65 | Минздрав РСФСР | |
| 204 | Фольга алюминиевая, кашированная клеями марок МЦП-IV (НМЗ), МЦП-IV (НВП) | Для упаковки сливочного масла | Церезин Полиизобутилен Канифоль Масло вазелиновое |
126-14/14063 25.09.69 | Минздрав СССР | |
| 205 | Фольга алюминиевая, кашированная клеем марки МЦП-III | Для упаковки сливочного масла | Парафин Полиизобутилен Канифоль Масло вазелиновое |
126-14/14063 25.09.69 | Минздрав СССР | |
| 206 | Фольга алюминиевая, кашированная клеем марки МЦП-V | Для упаковки сливочного масла | Парафин Церезин Полиизобутилен Масло вазелиновое |
126-14/14063 25.09.69 | Минздрав СССР | |
| 207 | Фольга, кашированная нестабилизированным полиэтиленом высокого давления марки П2070П | Для упаковки и выпуска опытной партии столовой горчицы и соуса «Южный» (расфасовка на автоматах «Волкогон», ФРГ) | 08с/Б-7б-154 02.02.68 | Минздрав РСФСР | ||
| 208 | Хохломская роспись | Ложки | Природная жирная глина; натуральное льняное масло; смесь подсолнечного натурального масла с лаком 4С (ГОСТ 5470-50); алюминиевый порошок-пудра (ГОСТ 5494-50); сажа газовая (ГОСТ 7848-56) или кадмий красный (ГОСТ 11826-66); лак 3-30-59 (СТУ 30-9131-63): ксиленол — фенолформальдегидная смола КЭ-Э (ксиленол, фенол, формальдегид), эпоксидная смола Э-05 (эпоксидная смола Э-33; дифенилолпропан, эпихлоргидрин, сульфат целлюлозы; пиперидин) |
08с/Б-7-713 20.06.69 | Минздрав РСФСР | |
| 209 | Церезин марок М-57, М-67 | Для пропитывания бумажных стаканчиков под расфасовку сметаны | 08с/Б-7-3157 14.12.64 | Минздрав РСФСР | ||
| 210 | Эмаль | Для покрытия внутренней поверхности бочек под тузлучные рыбопродукты | Канифоль (ГОСТ 797-50), парафин (ГОСТ 784-53), петролатум (ГОСТ 4096-62) | 123-8/80 29.08.57 | Минздрав СССР | |
| 211 | Эмаль белковоустойчивая ЭП-547 (с алюминием) <15> | Лакирование белой жести для консервной тары под мясные и рыбные продукты (натуральные и в масле) | Фенолпаратретбутил — фенолформальдегидная смола ФПФ-1: паратретбутилфенол, фенол, формальдегид, едкий натр, соляная кислота, бутанол, ортофосфорная кислота. Эпоксидная смола Э-05: дифенилолпропан, эпихлоргидрин, едкий натр, карбоксиметилцеллюлоза (КМЦ) | 126-14/17063 08.05.69 | Минздрав СССР | |
| 212 | Эмаль белковоустойчивая ЭП-547 (с окисью алюминия) <15> | Лакирование белой жести для консервной тары под мясные и рыбные продукты (натуральные и в масле) | Фенолпаратретбутил — фенолформальдегидная смола ФПФ-1: паратретбутилфенол, фенол, формальдегид, едкий натр, соляная кислота, бутанол, ортофосфорная кислота. Эпоксидная смола Э-05, дифенилолпропан, эпихлоргидрин, едкий натр, карбоксиметилцеллюлоза (КМЦ) | 126-14/18963 04.09.69 | Минздрав СССР | |
| 213 | Эмаль белковоустойчивая ЭП-527 (с окисью цинка) <15> | Лакирование белой жести для консервной тары под рыбные консервы (натуральные и в масле) | Фенолпаратретбутил — фенолформальдегидная смола ФПФ-1: паратретбутилфенол, фенол, формальдегид, едкий натр, соляная кислота, бутанол, ортофосфорная кислота. Эпоксидная смола Э-05: дифенилолпропан, эпихлоргидрин, едкий натр, карбоксиметилцеллюлоза (КМЦ). Алкидноэпоксидная смола Э-30К: а) смола Э-40: дифенилолпропан, эпихлоргидрин, едкий натр; б) смола полиэфирная ГФ-019: фталевый ангидрид, масло льняное, масло тунговое, глицерин, каустическая сода; в) тетралин (тетрагидронафталин) или ксилол, ортофосфорная кислота, окись цинка |
126-14/Г7063 08.05.69 | Минздрав СССР | |
| 214 | Эмаль белковоустойчивая КР-1 | ТУ МППТ 405-51 | 1. Для лакирования консервной тары для крабов | Смола 101: паратретичный бутилфенол, формальдегид, цинк-паста, масло тунговое, уайт-спирит, резинат марганца, скипидар | 08с/Б-7-446 11.05.62 | Минздрав РСФСР |
| 2. Для покрытия жестяной тары для кондитерских изделий, какао, кофе, чая | ||||||
| 215 | Эмаль бронзовая | Для окраски наружной поверхности утятниц | 08с/Б-7-2487 14.12.66 | Минздрав РСФСР |
| 216 | Эмаль ВХЭ-4001 | ВТУ МХП 4405-55 | Для покрытия внутренней поверхности хлеборазвозочных кузовов | 123-10/94 16.04.56 | Минздрав СССР | |
| 217 | Эмаль глифталевая А-170 | Для покрытия винодельческого оборудования | Состав: лак глифталевый, пудра алюминиевая (ГОСТ 5494-50) | 113-17/380 03.07.54 | Минздрав СССР | |
| 218 | Эмаль глифталевая алюминиевая АЛ-701 | ТУ МХП 1924-49 | Для покрытия тары под сливочное и топленое масло | Состав: глифталевый лак, алюминиевая пудра (ГОСТ 5494-50) | 113-17/380 03.07.54 | Минздрав СССР |
| 219 | Эмаль перхлорвиниловая марки ХВ-124 (ПХВ-715Т), ПФ-115 | ГОСТ 10144-62 ГОСТ 6465-63 | Для окраски станков и оборудования, не имеющих непосредственного контакта с пищевыми средами | 123-14/10977 08.09.65 | Минздрав СССР | |
| 220 | Эмаль пентафталевая серо-голубая N 560 (бессвинцовая эмаль) | ТУ МХП 1764-48 | Для покрытия наружных поверхностей хлебных автофургонов | 113-17/270 07.05.54 | Минздрав СССР | |
| 221 | Эмаль молотковая марки МЛ-165 | ГОСТ 12034-66 | Для окраски наружной поверхности овощерезки | 08с/Б-7-662 28.04.69 | Минздрав РСФСР | |
| 222 | Эмаль МЛ-123 | СТУ 14-39963-07 | Для покрытия только наружной поверхности хлебниц | 08с/Б-7-2562 14.01.69 | Минздрав РСФСР | |
| 223 | Эмаль МЛ-152 | МРТУ 6-10-64267 | То же | То же | Минздрав РСФСР | |
| 224 | Эмаль ХСЭ-А (ХС-76Т) | Для покрытия внутренней поверхности металлических и железобетонных резервуаров, предназначенных для хранения и обработки вина, пива и др. пищевых жидкостей | Состав: смола СВХ-40 (ВТУ 410-54), двуокись титана, растворитель Р-4 (ГОСТ 7827-55), ацетон (ГОСТ 2768-60), бутилацетат (ГОСТ 8981-59), толуол (ГОСТ 4809-49) | 123-7/1 30.01.62 | Минздрав СССР | |
| 225 | Эмаль Э-1 | В качестве верхнего слоя эмалевого покрытия труб для подачи забортной воды на крабовых заводах | Двуокись кремния, окись кальция, окись алюминия, кобальт, трехокись бора, двуокись титана, окись калия, натрий алюмофторид | 123-9/67 23.04.64 | Минздрав СССР | |
| 226 | Эмалевая пленка КР-1 | Для покрытия тары, предназначенной для хранения и транспортировки топленого масла | Смола 101: паратретичный бутилфенол, формальдегид: масло тунговое, уайт-спирит, резинат марганца, скипидар | 113-17-111 08.04.54 | Минздрав СССР | |
| 227 | Эмульсия поливинилацетатная водная | ГОСТ 10002-62 | 1. Для склеивания папирос | 123-11/12 13.02.58 | Минздрав СССР | |
| 2. Для склеивания бумажной тары (стаканчиков, коробок), предназначенной для упаковки пищевых продуктов при условии, если они не соприкасаются с клеем | 123-7/41 03.05.61 | Минздрав СССР | ||||
| 3. Для склеивания пакетов в автоматах марки «БРА» при условии, что эмульсия не будет соприкасаться с пищевыми продуктами | 08с/Б-7-1998 04.09.64 | Минздрав РСФСР | ||||
| 4. Для склеивания пакетов при условии: а) затаривание в пакеты сухих продуктов с содержанием влажности не более 15% б) склеивание должно проводиться таким образом, чтобы эмульсия не соприкасалась с пищевым продуктом |
08с/Б-7-2423 24.11.61 | Минздрав РСФСР | ||||
| 228 | Эмульсия поливинилацетатная водная марки ВВ | ГОСТ 10002-62 | Для изготовления кашированной фольги, для упаковки только сухих продуктов (содержащих влагу не более 15%) | 08с/Б-7-1263 11.08.64 | Минздрав РСФСР | |
| 229 | Эмульсия силиконового масла марки ПМС-400 | Вносится в рецептуру латексной уплотнительной пасты N 1 и N 2 в количестве 0,001% | 126-14/452-3 24.02.69 | Минздрав СССР | ||
| Импортные материалы | ||||||
| 1 | Бумага (Финляндия), соответствующая бумаге с полиэтиленовым покрытием | СТУ 36-10-63 | Пакеты «тетрапак» для упаковки сметаны 10% и 30% жирности | Бумага — основа для пакетов СТУ 30-6029-61, полиэтилен высокого давления нестабилизированный по МРТУ 6-05-889-64, группа Л-27, марка П2070, индекс расплава 6 — 7 | 123-11/49-7 03.04.65 | Минздрав СССР |
| 2 | Бумага, покрытая эмульсией «Диофан», эмульсия импортная (ФРГ) | 1. Тара для длительного хранения сыпучих пищевых продуктов | N 688 09.02.65 | Минздрав УССР | ||
| 2. Для хранения влажных и жирных продуктов в сроки, соответствующие требованиям государственных стандартов | ||||||
| 3 | Добавки в пластмассы (препараты английских фирм РЕКС Кембели Ко ЛДТ и Армар Хесс Кемикал ЛДТ) адоджен 58 и армид-0 | Для пищевой промышленности, водоснабжения, для изготовления детских игрушек | 126-5/483-3 13.12.68 | Минздрав СССР | ||
| 4 | Картон финский хромэрзац с проклейкой 1 верхнего и нижнего слоев | Коробки под упаковку мороженого «Пломбир» по 250 г | 08с/Б-7-1018 12.05.68 | Минздрав РСФСР | ||
| 5 | Лак МО 413-р 18388 (ГДР) | Для лакирования консервной тары с последующей стерилизацией | 123-8/22-7 30.01.65 | Минздрав СССР | ||
| 6 | Оболочка вискозная фирмы «ФА Калле АГ» (ФРГ) <16> | Упаковка сосисок при условии поставки фирмой стандартных партий оболочек (сравнимых) | 04-5/253 26.11.69 | Минздрав СССР | ||
| 7 | Паста уплотнительная «S505» японской фирмы «ТОЙОИНК» | Для герметизации швов жестяной тары в консервной промышленности | Синтетический каучук CBR, канифоль, камедь натуральная, метилцеллюлоза, сажа, окись цинка, глинозем, антиокислитель, диспергент, бензоат натрия, олеиновая кислота | 126-5/37-2 13.06.69 | Минздрав СССР | |
| 8 | Пластик твердый ПХВ (пластик фирмы «Штаурен», ФРГ) | Для изготовления упаковки сыра «Янтарь» | 126-11/960-3 04.10.68 | Минздрав СССР | ||
| 9 | Пленка импортная целлофан-полиэтиленовая | Для упаковки майонеза | 08с/Б-7-1284 26.07.66 | Минздрав РСФСР | ||
| 10 | Пленка «Крехалон» (типа «Саран») японской фирмы «Куреха» 3 типов: «FD» толщиной 0,35 мм; «ЕД» и «СА» толщиной 0,40 мм | Упаковка пищевых продуктов. Хранение при температуре не выше 18 град. C | 04-5/8 15.01.68 | Минздрав СССР | ||
| 11 | Пленка итальянская комбинированная фирмы «Грейс» <17> | Упаковка рыбной продукции при условии строгого соблюдения «Правил упаковки рыбной продукции и рыбной кулинарии в пакеты и мешки из синтетических пленок», разработанных Минрыбхозом СССР и согласованных с Минздравом СССР | Рецепт 1226: нейлон, полиэтилен Рецепт 1515: полиэтилен, лакированный целлофан с двух сторон |
126-14/1083/ 3 09.09.69 | Минздрав СССР | |
| 12 | Пленка японская «Эстлон-HP» фирмы «ХОКУЕ БУССАН КАЙСЯ» | Для расфасовки рыбной продукции (кулинария, копченые, соленые изделия, прессервы) | 08с/Б-7б1864 19.05.69 | Минздрав РСФСР | ||
| 13 | Пленка итальянская комбинированная «Дарван» из полиэтилен-целлофана | Для упаровки рыбной продукции: 1. Балык из осетровых рыб холодного копчения 2. Балык из дальневосточных лососевых холодного копчения 3. Лососи соленые дальневосточные 4. Лососи соленые |
126-14/22-3 10.04.69 | Минздрав СССР | ||
| 14 | Пленки поливинилхлоридные фирмы «Фолиен фабрик форгейм» | Упаковка сахароварных продуктов на автомате «Хассия» | 126-14/51-3 17.10.69 | Минздрав СССР | ||
| 15 | Пленка «Саран» (США) | Для упаковки бескоркового сыра | Продукт полимеризации винилденхлорида и хлористого винила | 123-9/134 19.08.64 | Минздрав СССР | |
| 16 | Пленка поливинилхлоридная марки VK 5911В и покровная поливинилхлоридная пленка марки HQ (Н913) фирмы «Калле» (ФРГ) | Изготовление коробочек емкостью 50, 150, 250 г для затаривания повидла <18> | 126-14/46-3 15.03.68 | Минздрав СССР | ||
| 17 | Покрытия лаковые импортные по образцам 9, 10 | Для покрытия фольги, предназначенной для упаковки пищевых продуктов при условии отсутствия непосредственного контакта окрашенной поверхности с пищевым продуктом | 126-22/896-3 27.03.68 | Минздрав СССР | ||
| 18 | Поливинилхлорид по рецептуре П-72 (импортный) | Для упаковки джемов, варенья (коробочки весом не менее 200 г) | Поливинилхлорид (с-60), сополимер (ПХ-2ЭГА), п. 601-17, полигард, CaO6, стеарат кальция, стеарат цинка, глицерин, диоктилфталат, эпоксидированное соевое масло, тинопал, двуокись титана | 126-5/398-3 09.01.69 | Минздрав СССР | |
| 19 | Поливинилхлорид марки П-73 | -«- | Поливинилхлорид (с-60), полигард, CaO6, стеарат кальция, стеарат цинка, глицерин, диоктилфталат, эпоксидированное соевое масло, тинопал | 126-5/398-3 09.01.69 | Минздрав СССР | |
| 20 | Полистирол японский марки «АсахиДау» 475-Д | Изготовление тары (стаканчики, коробочки) для упаковки плавленых сыров <19> | 126-5/195-3 23.07.69 | Минздрав СССР | ||
| 21 | Полистирол марки 475-К японской фирмы «Асахи-Дау» | Для внутренней облицовки холодильников | 08с/Б-7-1021 30.06.66 | Минздрав РСФСР | ||
| 22 | Полистирол ударопрочный марки 475 японской фирмы «Асахи-Дау» | Для внутренней облицовки домашних холодильников «Бирюса» | 126-9/869-3 13.09.67 | Минздрав СССР | ||
| 23 | Полистирол ударопрочный марки 475-К фирмы БАСФ (ФРГ) | Для производства домашних холодильников «Днепр» | 126-5/267а-3 19.09.67 | Минздрав СССР | ||
| 24 | Полистирол марки 475-К фирмы БАСФ (ФРГ) | Для изготовления тазиков для созревания мяса | 08с/Б-7б-608 13.05.69 | Минздрав РСФСР | ||
| 25 | Полиэтилен (Финляндия) | Для изготовления вкладышей для упаковки маргарина | 123-11/74-7 17.06.65 | Минздрав СССР | ||
| 26 | Полиэтилен низкой плотности («Алкатен», Англия) | Для упаковки мясных продуктов | 123-7/32 14.04.61 | Минздрав СССР | ||
| 27 | Полиэтилен высокого давления марки «Супротен» | Для упаковки хлеба для дительного хранения вторым слоем после фильтровальной бумаги | 123-10/22 28.07.60 | Минздрав СССР | ||
| 28 | Полиэтилен низкой плотности ДГД 2005 (итальянская фирма «Келене») | Крышки для укупоривания стеклянных банок | N 716 12.02.65 | Минздрав УССР | ||
| 29 | Полиэтилен низкой плотности (пленка) «Супротен» (ФРГ) | Для тары, предназначенной предохранять хлеб и плоды от усушки | 123-10/51 10.12.60 | Минздрав СССР | ||
| 30 | Полиэтиленовые мешки фирмы «Мейк Трейдинг Ко ЛТД» | Для затаривания сухих сыпучих продуктов (макаронные изделия, сахар и др.) | 08с/Б-7-3113 03.12.64 | Минздрав РСФСР | ||
| 31 | Прокладки N 2 из натуральной пробки (Алжир) | Для укупорки фруктовых вод и пива | 126-14/905-3 27.06.69 | Минздрав СССР | ||
| 32 | Прокладки пробковые прессованные марокканской фирмы «Аглорекс» и алжирской фирмы «Интермундо» | Укупорка пива и безалкогольных напитков | 126-14/97-3 01.03.68 | Минздрав СССР | ||
| 33 | Смола эпоксидная «Эпросин» (ЧССР) | Для покрытия емкостей, имеющих непосредственный контакт с вином | 123-7/38 29.05.61 | Минздрав СССР |
<1> При условии, если клей будет готовиться в изолированном помещении.
<2> Временно, до 01.01.1972.
<3> В опытном порядке.
<4> Хранение продуктов при температуре не выше 20 град. C. Затаривание может производиться при температуре 60 — 80 град. C.
<5> Упаковку производить без вакуума.
<6> Временно, количество 5 — 30 резервуаров.
<7> В порядке опытной эксплуатации.
<8> Применять свежеприготовленные растворы.
<9> В инструкции по применению необходимо оговорить тщательную обработку деталей горячей водой перед первым употреблением.
<10> Использовать полимеры светлых тонов.
<11> В технологической инструкции предусмотреть пропускание через анионит и катионит до начала фильтрации раствора агароида горячей (80 — 90 град. C) воды в количестве не менее 20-кратного к весу ионитов для обеспечения не только их прогревания, но и промывания.
<12> Временно на два года (1970 — 1971).
<13> При условии укупорки крышками мгновенно при термической обработке (стерилизации) содержимого банки.
<14> Временно, на 2 года.
<15> До 01.01.1972.
<16> Временно.
<17> Пищевой продукт контактирует с полиэтиленовой поверхностью при температуре не выше 8 град. C. Сосиски рыбные в течение 12 часов, рыба жареная — 48 часов.
<18> Срок хранения не более 3 — 5 месяцев.
<19> При условии одноразового замачивания тары в горячей воде при температуре 60 град. C и выдерживания в течение 24 часов с последующей промывкой проточной водой.
Отбор проб воздуха
Атмосферный воздух
Промышленные выбросы
Воздух рабочей зоны
Воздух замкнутых помещений
Отбор проб атмосферного воздуха
| № | № НД | Название определяемой характеристики | На что отбирать | Скорость отбора | Время отбора | Срок хранения | Примечание | ||||||||
| 1 | ГОСТ 17.2.4-05 | взвешенные вещва (пыль) | фильтр АФА-ВП | 75 дм3/мин | По окончании отбора регистрируют общий объем пропущенного воздуха. | — | — | ||||||||
| 2 | РД 52.04.792-14 (ФР.1.31.2015.19877) | Азота диоксид | двух последовательно соединенных СТ 412 и расположенный между ними осушитель и СТ-окислитель | 0,5дм3/мин | 30 | 10 дней | При определении разовой концентрации оксида азота и диоксида азота: присоединяют систему, состоящую из двух последовательно соединенных СТ 412 и расположенных между ними по ходу воздуха: колонки, заполненной 10 см осушителя, и СТ-окислителя. При отборе проб колонка и сорбционные трубки должны быть укреплены в вертикальном положении слоем сорбента вниз. Воздух должен проходить снизу вверх. Все соединения делаются резиновыми муфтами встык. При определении суточной концентрации оксида азота и диоксида азота из одной пробы исследуемый воздух аспирируют дискретно, двенадцать раз в 1 сут по 20 мин через равные промежутки времени с расходом 0,3 дм/мин через систему, состоящую из двух последовательно соединенных СТ 412, подготовленных к отбору и расположенных между ними по ходу воздуха: колонки, заполненной 100 см осушителя, и СТ-окислителя.При отборе проб колонки и сорбционные трубки должны быть укреплены в вертикальном положении слоем сорбента вниз. Воздух должен проходить снизу вверх. Все соединения делаются резиновыми муфтами встык. Отбор проб можно проводить при температуре воздуха, проходящего через сорбционную трубку, от минус 10°С до 40°С. При отсутствии сорбционных трубок СТ 412 вместо каждой из них в схему включают по две сорбционные трубки СТ 212. |
||||||||
| Азота оксид | |||||||||||||||
| 3 | РД 52.04.791-14 (ФР.1.31.2015.19887) | Аммиак | сорбционна трубка | 2,0 дм3/мин | 20 | 5 дней | Сорбционная трубка должна быть укреплена в вертикальном положении слоем сорбента вниз.С целью задержки аэрозоля солей аммония при отборе проб к сорбционной трубке на входе воздуха подсоединяют фильтродержатель с фильтром АФА-ВП-10. Все соединительные трубки должны быть изготовлены из фторопласта и соединяться встык чистыми (промытыми и высушенными) резиновыми муфтами. Необходимо тщательно следить за тем, чтобы внутренняя поверхность соединительных трубок была сухой во избежание сорбции аммиака в них. Воздух должен проходить снизу вверх. Отбор проб можно проводить при температуре воздуха, проходящего через сорбционную трубку, от 0°С до 40°С. |
||||||||
| 4 | МУК 4.1.596-96 | Аммоний сернокислый (по иону аммония) | АФА или «синяя лента» | 20 дм3/мин | 10-20 | 7 дней | — | ||||||||
| Аммоний надсернокислый (по иону аммония) | 1 день | ||||||||||||||
| 5 | РД 52.04.797-14 (ФР.1.31.2015.19878) | Водород фтористый (гидрофторид) | сорбционна трубка | 3 дм3/мин | 20 | 7 дней | При отборе сорбционная трубка должна быть укреплена в вертикальном положении, слоем сорбента вниз. Воздух должен идти снизу вверх. Отбор проб можно проводить при температуре анализируемого воздуха от минус 10°С до 40°С. Перед сорбционной трубкой с помощью полиэтиленового уголка устанавливают фильтродержатель с фильтром для улавливания твердых фторидов и других аэрозолей. Сорбционная трубка, уголок и фильтродержатель должны быть соединены встык, чтобы не было контакта воздуха с резиной.При определении суточных концентраций отбирают не менее четырех разовых проб через равные промежутки времени. | ||||||||
| 6 | РД 52.04.793-14 (ФР.1.31.2015.19882) | Водород фтористый (гидрофторид) | фильтр АФА-ПВ-10 и сорбционная трубка | 4 дм3/мин | 20 | 10 дней | Фильтродержатель с фильтром и сорбционная трубка соединяются с помощью резиновой муфты встык. Сорбционная трубка должна быть укреплена в вертикальном положении слоем сорбента вниз. Воздух должен поступать снизу вверх. Отбор проб можно проводить при температуре анализируемого воздуха, проходящего через сорбционную трубку, от 0°С до 40°С. При ожидаемых концентрациях хлорида водорода от 5 до 10 ПДК объемный расход воздуха следует снизить до 2 дм/мин. При определении суточной концентрации отбирают не менее четырех разовых проб через равные промежутки времени. |
||||||||
| 7 | РД 52.04.795-14 (ФР.1.31.2015.19886) | Сероводород (дигидросульфид) | сорбционная трубка | 4 дм3/мин | 20 | 2-3 дня | При отборе сорбционная трубка должна быть укреплена в вертикальном положении слоем сорбента вниз и защищена от света экраном из черной бумаги или фольги. | ||||||||
| 8 | РД 52.04.794-14 (ФР.1.31.2015.19884) | Серы диоксид | поглотители Рихтера | 2,5 дм3/мин | 20 | 7 дней | При ожидаемых больших массовых концентрациях диоксида серы расход воздуха может быть снижен до 0,5 дм/мин.При определении суточных концентраций отбирают не менее четырех разовых проб через равные промежутки времени. Отбор проб можно проводить при температуре анализируемого воздуха от 0°С до 40°С.Пробы в процессе отбора и при хранении необходимо защищать от прямого солнечного света. | ||||||||
| 9 | РД 52.04.831-15 (ФР.1.31.2016.23390) | Сажа (углеродосодержащий аэрозоль) | АФА-ХП-10 АФА-ВП10 | 20 дм3/мин | 30 | неограничен | Отбор проб проводится с наветренной стороны на высоте 1,5 м от поверхности земли. | ||||||||
| 10 | РД 52.04.798-14 (ФР.1.31.2015.19880) | хлор | поглотитель Рихтера | 0,5 дм3/мин | 20 | 3 дня | При определении суточной концентрации отбирают не менее четырех разовых проб через равные промежутки времени. | ||||||||
| 11 | Мук 4.1.615-96 | Хлорид калия | АФА-ВП-20 | 10 дм3/мин | 12.5 | 1 месяц | — | ||||||||
| 12 | МУК 4.1.1044а-01 | Диметиамин | одновременно на 2 сорбционные трубки (сорбент Chromosorb 106 или 103) | 0,2 дм3/мин | 10 | 3 дня | Воздух аспирируют через предварительно охлажденное до 0°С отверсие сорбционной трубки. | ||||||||
| Диэтиламины | |||||||||||||||
| Пропиламины | |||||||||||||||
| Триэтиламины | |||||||||||||||
| Этиламины | |||||||||||||||
| Диметилформамид | |||||||||||||||
| 13 | ГОСТ Р ИСО 16017-1 | Ацетонитрил | сорбционная трубка-концентратор (сорбент TENAX) | (1-10) дм3/мин | В зависимости от условий | 8 часов ( или сорбционные трубки помещают в чистый без покрытия охлаждаемй герметичный контейнер. | — | ||||||||
| Акрилонитрил (проп-2-енонитрил) | |||||||||||||||
| Гексан | |||||||||||||||
| Гептан | |||||||||||||||
| Декан | |||||||||||||||
| Додекан | |||||||||||||||
| Нонан | |||||||||||||||
| Октан | |||||||||||||||
| Углеводороды алифатические предельные бензиновой фракции С6-С12 | |||||||||||||||
| Ундекан | |||||||||||||||
| Ацетон (пропан-2-он) | |||||||||||||||
| Метилэтилкетон (Бутанон) | |||||||||||||||
| Гексанон | |||||||||||||||
| Диацетоновый спирт (4-гидрокси-4-метил-2-пентанон) | |||||||||||||||
| Циклогексанон | |||||||||||||||
| Бензол | |||||||||||||||
| Изопрпилбензол (Кумол) | |||||||||||||||
| Ксилол (диметилбензол) | |||||||||||||||
| Псевдокумол (1,2,4- триметилбензол) | |||||||||||||||
| Стирол (этенилбензол) | |||||||||||||||
| Толуол (метилбензол) | |||||||||||||||
| Этилбензол | |||||||||||||||
| Винилхлорид (хлорэтен) | |||||||||||||||
| Дихлорметан | |||||||||||||||
| 1.1-дихлорэтилен | |||||||||||||||
| Трихлорэтан | |||||||||||||||
| Трихлорэтилен | |||||||||||||||
| Четыреххлористый углерод (тетрахлорметан) | |||||||||||||||
| Хлорбензол | |||||||||||||||
| Хлороформ (трихлорметан) | |||||||||||||||
| Бутанол | |||||||||||||||
| 1.2-дихлорэтан | |||||||||||||||
| ГОСТ Р ИСО 16017-1 | Изобутанол | сорбционная трубка-концентратор (сорбент TENAX) | (1-10) дм3/мин | В зависимости от условий | 8 часов ( или сорбционные трубки помещают в чистый без покрытия охлаждаемй герметичный контейнер. | — | |||||||||
| (2-метилпропан-1-ол) | |||||||||||||||
| Метанол (карбинол) | |||||||||||||||
| Амиловый спирт (пентанол) | |||||||||||||||
| Пропиловый спирт (прпан-1-ол) | |||||||||||||||
| Циклогексанол (циклогексиловый спирт) | |||||||||||||||
| Этанол (этиловый спирт) | |||||||||||||||
| Фенол (гидроксибензол) | |||||||||||||||
| Бутилацетат | |||||||||||||||
| Дибутилфталат | |||||||||||||||
| Диметилфталат | |||||||||||||||
| Демитилизофталат | |||||||||||||||
| Диэтилфталат | |||||||||||||||
| Изобутилацетат | |||||||||||||||
| Метилацетат | |||||||||||||||
| Пропилацетат | |||||||||||||||
| Винилацетат (Этенилацетат) | |||||||||||||||
| Этилакрилат | |||||||||||||||
| Этилацетат | |||||||||||||||
| 14 | МУК 4.1.629-96 | Нитрилы (С10-С16) | поглотительный прибор Яворовской | 10 дм3/мин | 30 | 5 часов | — | ||||||||
| 15 | ФР.1.31.2011.11272 (М-22) | Дивинил (бута-1,3-диен) | 2 последовательно соединенные (встык) сорбционне трубки-концентратора (сорбент TENAX) | 0,3 дм3/мин | 30 | 2 дня в холодильнике | При определении приземной концентрации вещества в атмосфере отбор проб проводятся на высоте 1,0 — 3,5 м от поверхности земли. Каждую пробу отбирают на 2 последовательно соединенные (встык) сорбционные трубки, установленные вертикально, входным отверстием вниз. При ожидаемых массовых концентрациях, не превышающих 1 мг/м3, отбор проб осуществляют с объемным расходом 0,3 дм3/мин в течение 30 минут. Если ожидаемые массовые концентрации превышают 1 мг/м3, отбирают несколько проб (2 — 3) с меньшим объёмным расходом в течение этого временного промежутка. |
||||||||
| Бензальдегид | |||||||||||||||
| Метилацетат | |||||||||||||||
| Изопропилацетат (1-метиэтилацетат) | |||||||||||||||
| н-прпилацетат | |||||||||||||||
| изобутилацетат | |||||||||||||||
| Н-Бутилацетат | |||||||||||||||
| Н-Амилацетат | |||||||||||||||
| Изопрен | |||||||||||||||
| Н-гексанол | |||||||||||||||
| 2-этилгексанол | |||||||||||||||
| октан-1-ол (н-октиловый спирт) | |||||||||||||||
| Бензиловый спирт (бензилкарбинол) | |||||||||||||||
| Пропионовая кислота | |||||||||||||||
| Пентановая к-та (валериановая) | |||||||||||||||
| Гексановая к-та (капроновая) | |||||||||||||||
| 2-этоксиэтиловый эфир уксусной к-ты | |||||||||||||||
| ФР.1.31.2011.11272 (М-22) | Метилцеллозольв (2-метаксиэтанол) | 2 последовательно соединенные (встык) сорбционне трубки-концентратора (сорбент TENAX) | 0,3 дм3/мин | 30 | 2 дня в холодильнике | При определении приземной концентрации вещества в атмосфере отбор проб проводятся на высоте 1,0 — 3,5 м от поверхности земли. Каждую пробу отбирают на 2 последовательно соединенные (встык) сорбционные трубки, установленные вертикально, входным отверстием вниз. При ожидаемых массовых концентрациях, не превышающих 1 мг/м3, отбор проб осуществляют с объемным расходом 0,3 дм3/мин в течение 30 минут. Если ожидаемые массовые концентрации превышают 1 мг/м3, отбирают несколько проб (2 — 3) с меньшим объёмным расходом в течение этого временного промежутка. |
|||||||||
| Изопрпилцеллозольв (2-изопропоксиэтанол) | |||||||||||||||
| Дифениловый эфир | |||||||||||||||
| Бутилцеллозольв (2-бутоксиэтанол) | |||||||||||||||
| Дурол (1,2,4,5-тетраметилбензол) | |||||||||||||||
| 1-метоксипрпопан-2-ол | |||||||||||||||
| 1-этоксипрпан-2-ол | |||||||||||||||
| 4-метилпентан-2-ол | |||||||||||||||
| Циклогексан | |||||||||||||||
| Метилбутонат | |||||||||||||||
| Этилбутаноат | |||||||||||||||
| Метилпропионат | |||||||||||||||
| Этилпропионат | |||||||||||||||
| Этиленгликоль (1,2-этандиол) | |||||||||||||||
| Пропиленгликоль (1,2-пропандиол) | |||||||||||||||
| 16 | ПНД Ф 13.1:2:3.23-98 (ФР.1.31.2015.20483) | бутены (бутен-1, бутен-2, изобутен) | шприцы | 7-10 кратная прокачка шприца | — | 5 часов | — | ||||||||
| Этен (этилен) | |||||||||||||||
| Пропен (пропилен) | |||||||||||||||
| С1-С5 (пред.угл) | |||||||||||||||
| 17 | ПНД Ф 13.1:2:3.25-99 (ФР.1.31.2015.20480) | С2-С5 (непредельные) (суммарно); | шприцы | 7-10 кратная прокачка шприца | — | 5 часов | — | ||||||||
| С1-С10 (предельные) (суммарно) | |||||||||||||||
| Бензол | |||||||||||||||
| Толуол | |||||||||||||||
| Этилбензол | |||||||||||||||
| Ксилолы | |||||||||||||||
| Стирол | |||||||||||||||
| 18 | ФР.1.31.2011.11269 (М-24) | изоционаты и ароматические амины (анилин, н-нитроанилин, толуилендиизоционат) | сорбционне трубки | 5 дм3/мин | 60 | — | Собирают 2 линии пробоотбора. В каждой линии по 2 оследовательно соединенне трубки, установленные вертикально сорбентом вниз. | ||||||||
| 19 | ПНД Ф 13.1:2:3.59-07 (ФР.1.31.2013.16458) (М 01-05) | углеводороды алифатические предельные керосиновой фракции С12-С19 | одноразовый пробоотбоник типа «карбон» | 0,2-0,3 дм3/мин | 20-30 | 7 дней в холодильнике | — | ||||||||
| 20 | МУК 4.1.1957-05 | винилхлорид | одновременно на 2 сорбционные трубки (сорбент Chromosorb 104) | 0,2 дм3/мин | 10 | 3 дня в холодильнике | Каждая проба воздуха одновременно отбирается на две сорбционные трубки с помощью 2 пробоотборников. Воздух прокачивают через узкое отверстие сорбционной трубки, охлаждаемой холодильником со льдом до 0 °С. |
||||||||
| ацетальдегид | |||||||||||||||
| 21 | МУК 4.1.616-96 | муравьиная к-та | сорбционне трубки СТ 212 | 5 дм3/мин | 20 | 10 дней | — | ||||||||
| уксусная к-та | |||||||||||||||
| пропионовая к-та | |||||||||||||||
| маслянная к-таа | |||||||||||||||
| валериановая к-та | |||||||||||||||
| капроновая к-та | |||||||||||||||
| 22 | ПНД Ф 13.1:3.68-09 (ФР.1.31.2015.19226) | Бензол | Поглотительный прибор с пористой пластиной | 0,5 дм/мин. | 20-30 | 3 дня в холодильнике | — | ||||||||
| Ксилол (диметилбензол) | |||||||||||||||
| 23 | МУК 4.1.632-96 | Этилтолуол | Поглотительный прибор с пористой пластиной | 0,5 дм/мин. | 30 | 1 месяц в темном и прохладном месте | — | ||||||||
| Пропилбензол | |||||||||||||||
| Псевдокумол (1,2,4- триметилбензол) | |||||||||||||||
| Нафталин | 2 последоватеьн соединенных поглотителя Рихтера | 3 дм3/мин | 25 | Для улавливания паров уксусной кислоты перед аспиратором помещают стеклянную трубку диаметром 2 см и длиной 10 см, заполненную гидроксидом калия. После отбора пробы концы поглотительных сосудов фиксируют стеклянными заглушками. Срок хранения пробы в пробирках с притертыми пробками в темном и прохладном месте — 1 месяц. | |||||||||||
| 24 | МУК 4.1.619-96 | Метилмеркаптан | сорбционная трубка-концентратор (сорбент TENAX) | 1,5 дм3/мин | 30 | 3 дня в холодильнике | — | ||||||||
| Пропилмеркаптан | |||||||||||||||
| Бутилмеркаптан | |||||||||||||||
| Смесь природных меркаптанов | |||||||||||||||
| 25 | РД 52.04.799-2014 (ФР.1.31.2015.19883) | Фенол (гидроксибензол) | сорбционна трубка СТ212 или СТ 412 | 5 дм3/мин | 30 | 2 дня при температуре от 18°С до 25°С в темноте | — | ||||||||
| 26 | МУК 4.1.1143-02 | Тиметоксам | фильтр «синяя лента» | 4 дм3/мин | 5 | в морозильной камере при температуре -12 °С в течение 30 дней, в холодильнике при 4 — 6 °С — в течение 7 дней. | — | ||||||||
| 27 | ПНД Ф 13.1:2:3.71-11 (ФР.1.31.2015.21767) | Алюминий | АФА-ВП; АФА-ХП; АФА-ХА | 50 дм3/мин | 20 | 1 месяц | Отбор проб осуществляется на высоте 1,5 метра от поверхности земли, фильтродержатель должен быть ориентирован навстречу ветровому потоку. | ||||||||
| Барий | |||||||||||||||
| Бериллий | |||||||||||||||
| Ванадий | |||||||||||||||
| Висмут | |||||||||||||||
| Вольфрам | |||||||||||||||
| Железо | |||||||||||||||
| Галлий | |||||||||||||||
| Кадмий | |||||||||||||||
| Кобальт | |||||||||||||||
| Кремний | |||||||||||||||
| Литий | |||||||||||||||
| ПНД Ф 13.1:2:3.71-11 (ФР.1.31.2015.21767) | Магний | АФА-ВП; АФА-ХП; АФА-ХА | 50 дм3/мин | 20 | 1 месяц | Отбор проб осуществляется на высоте 1,5 метра от поверхности земли, фильтродержатель должен быть ориентирован навстречу ветровому потоку. | |||||||||
| Марганец | |||||||||||||||
| Мышьяк | |||||||||||||||
| Медь | |||||||||||||||
| Молибден | |||||||||||||||
| Серебро | |||||||||||||||
| Никель | |||||||||||||||
| Олово | |||||||||||||||
| Ртуть | |||||||||||||||
| Свинец | |||||||||||||||
| Селен | |||||||||||||||
| Сурьма | |||||||||||||||
| Титан | |||||||||||||||
| Хром | |||||||||||||||
| Цинк | |||||||||||||||
| 28 | РД 52.04.823-2015 (ФР.1.31.2016.23399) | Формальдегид | Поглотительный прибор Рыхтера | 2 дм3/мин | 30 | 5 дней | — |
Отбор проб промышленных выбросов
| № | № НД | Название определяемой характеристики | На что отбирать | Скорость отбора | Время отбора | Срок хранения | Примечание | ||||||||
| 1 | ГОСТ 33007 | Запыленность (массовое содержание взв. Частицы) | фильтр АФА-ВП | 10-20 дм3/мин | — | — | — | ||||||||
| 2 | ПНД Ф 13.1.33-02 (ФР.1.31.2014.18977) | Аммиак | 2 последовательно соед-ых поглотителя с пористой пластинкой | 0,5 дм3/мин | — | — | — | ||||||||
| 3 | М-20 (ФР.1.31.2011.11274) | Неорганические соединения фосфора (V) (в пересчете на дифосфор пентооксид) | Сдвоенный фильтр АФА-ВП(стеклянная пробоотборная трубка с нужным диаметром носика) | 20 дм3/мин | 20 | 3 дня | — | ||||||||
| 4 | М-7 (ФР.1.31.2011.11266) | Аэрозоль едких щелочей (по гидроксиду натрия) | Сдвоенный фильтр АФА-ВП(стеклянная пробоотборная трубка с нужным диаметром носика) | 20 дм3/мин | 20 | 3 дня | — | ||||||||
| 5 | ПНД Ф 13.1.45-03 (ФР.1.31.2015.19221) | Водород фтористый (гидрофторид) | 3 последовательно соединенных поглотителя Рихтера | (12-16) дм3/мин | — | — | Чтобы исключить поглощение фтористого водорода резиновым шлангом, поглотительные приборы, пробоотборная трубка и ловушка соединяются встык. Патроны предварительно набивают фторопластовой стружкой, а затем вставляют тампон из волокна фторин. Для сухих газов с температурой менее 80°С используют полиэтиленовые патроны, более 80°С — фторопластовые патроны. При отборе проб фтористого водорода из влажных газов используют обогреваемый патрон. Подогрев применяется для предотвращения поглощения фтористого водорода влагой, конденсирующейся в патроне. Расход газа при отборе 0,5-2,5 дм/мин и разряжение 3,3-26,6 кПа. Температура внутри патрона должна быть 160-180°С. | ||||||||
| 6 | ПНД Ф 13.1.42-03 (ФР.1.31.2015.19224) | Водород хлористый (гидрохлорид) | 2 последовательно соед-ых поглотителя с пористой стеклянной пластинкой | 1 дм3/мин | 10-30 | 1 день | — | ||||||||
| 7 | М-3 (ФР.1.31.2011.11281) | Кислота серная (аэрозоль) | Сдвоенный фильтр АФА-ВП или АФА-ХП(стеклянная пробоотборная трубка с нужным диаметром носика) | 20 дм3/мин | 20 | 2 дня | — | ||||||||
| 8 | ПНД Ф 13.1.46-04 (ФР.1.31.2007.03828) | Пары серной к-ты и аэрозоли триоксида серы (в пересчете на серную кислоту) | Стеклянная пробоотборная кассета | (0,5-2,0) дм3/мин | — | — | — | ||||||||
| 9 | ПНД Ф 13.1.34-02 (ФР.1.31.2007.03824) | Сероводород (дигидросульфид) | 4 последовательно соединеннх поглотителя Рихтера | 0,4 дм3/мин | 10-30 мин | 1 день (консервация 0,001% р-ром рутина: 5-7 дней) | По окончании пробоотбора необходимо определить конечную концентрацию щелочи в поглотительном растворе, которая должна быть не менее 2 моль/дм³. Для этого 1 см³ раствора гидроксида натрия С(NaOH) = 5 моль/дм³ титруют раствором соляной кислоты С(НСl) = 0,5 моль/дм³ в присутствии 1 — 2 капель фенолфталеина до исчезновения розовой окраски в течение 1 минуты. Фиксируют объем соляной кислоты, пошедший на титрование (V1). К бесцветному раствору добавляют 1 каплю метилоранжа (раствор желтый) и продолжают титровать раствором соляной кислоты до перехода от желтой окраски к оранжевой. Фиксируют объем соляной кислоты, пошедший на титрование щелочного раствора с метилоранжем. Концентрацию гидроксида натрия (моль/дм³) рассчитывают по формуле: C(NaOH) = ((V1 — V2) * C(HCl))/V3 где V1 — количество соляной кислоты, пошедшее на титрование пробы щелочного раствора с индикатором фенолфталеином, см³; V2 — количество соляной кислоты, пошедшее на титрование пробы щелочного раствора с индикатором метилоранжем, см³; С(HCl) — массовая концентрация соляной кислоты, С(HCl) = 0,5 моль/дм³; V3 — объем пробы щелочного раствора, отобранный для анализа, см³. Если конечная концентрация щелочи после отбора проб составляет менее 2 моль/дм³, то пробоотбор повторяют с использованием поглотительного раствора с концентрацией щелочи 8 моль/дм³. После пропускания порции газа поглотители отсоединяют от системы и закрывают свободные отростки заглушками. Пробы анализируют в день отбора или консервируют 0,001 %-ным раствором рутина (арабино-галактана) на 5 — 7 дней. |
||||||||
| Метилмеркаптан (метантиол) | |||||||||||||||
| 10 | ФР.1.31.2001.00384 | Углерод (сажа) | АФА-ВП-20, АФА-ВП-10 | (1-св.40) дм3/мин | 20 | — | — | ||||||||
| 11 | ПНД Ф 13.1.50-06 (ФР.1.31.2015.19220) | Хлор | Поглотительнй прибор Зайцева | 0,25 дм3/мин | 20 | 1 день | При отборе проб на прямом свету поглотитеьнй прибор оборачивают черной бумагой | ||||||||
| 12 | ФР.1.31.2011.11265 (М-10) | акрилонитрил (пропан-2-енонитрил) | сорбционная трубка-концентратор (сорбент TENAX) | 10 дм3/мин | 20 | Могут храниться в холодильнике 2 недели | Не менее 3-х проб (по 2-м параллельным в каждой). | ||||||||
| Ацетальдегид (Альдегид уксусный, этаналь) | 5 дм3/мин | Сразу помещается в охлажденный контейнер и хранится не более 2 дней | |||||||||||||
| Дихлорметан (Мителенхлорид) | 10 дм3/мин | Могут храниться в холодильнике 2 недели | |||||||||||||
| Дихлорэтан | |||||||||||||||
| Тетрохлорэтилен (перхлорэтилен) | |||||||||||||||
| Трихлорэтилен (трихлорэтен) | |||||||||||||||
| Четвреххлористый углерод (тетрахлорметан) | |||||||||||||||
| Хлороформ (трихлорметан) | |||||||||||||||
| Эпихлоргидрин (хлорметилоксиран) | |||||||||||||||
| 13 | ФР.1.31.2011.11272 (М-22) | Дивинил (бута-1,3-диен) | сорбционная трубка-концентратор (сорбент TENAX) | 0,1 дм3/мин | 20 | 7 дней в холодильнике | Производят 1-3 последовательных отбора проб (по 2 параллельных пробы для каждого отбора) | ||||||||
| Бензальдегид | |||||||||||||||
| Метилацетат | |||||||||||||||
| Изопропилацетат (1-метиэтилацетат) | |||||||||||||||
| н-прпилацетат | |||||||||||||||
| изобутилацетат | |||||||||||||||
| Н-Бутилацетат | |||||||||||||||
| Н-Амилацетат | |||||||||||||||
| Изопрен | |||||||||||||||
| Н-гексанол | |||||||||||||||
| 2-этилгексанол | |||||||||||||||
| октан-1-ол (н-октиловый спирт) | |||||||||||||||
| Бензиловый спирт (бензилкарбинол) | |||||||||||||||
| Пропионовая кислота | |||||||||||||||
| ФР.1.31.2011.11272 (М-22) | Пентановая к-та (валериановая) | сорбционная трубка-концентратор (сорбент TENAX) | 0,1 дм3/мин | 20 | 7 дней в холодильнике | Производят 1-3 последовательных отбора проб (по 2 параллельных пробы для каждого отбора) | |||||||||
| Гексановая к-та (капроновая) | |||||||||||||||
| 2-этоксиэтиловый эфир уксусной к-ты | |||||||||||||||
| Метилцеллозольв (2-метаксиэтанол) | |||||||||||||||
| Изопрпилцеллозольв (2-изопропоксиэтанол) | |||||||||||||||
| Дифениловый эфир | |||||||||||||||
| Бутилцеллозольв (2-бутоксиэтанол) | |||||||||||||||
| Дурол (1,2,4,5-тетраметилбензол) | |||||||||||||||
| 1-метоксипрпопан-2-ол | |||||||||||||||
| 1-этоксипрпан-2-ол | |||||||||||||||
| 4-метилпентан-2-ол | |||||||||||||||
| Циклогексан | |||||||||||||||
| Метилбутонат | |||||||||||||||
| Этилбутаноат | |||||||||||||||
| Метилпропионат | |||||||||||||||
| Этилпропионат | |||||||||||||||
| Этиленгликоль (1,2-этандиол) | |||||||||||||||
| Пропиленгликоль (1,2-пропандиол) | |||||||||||||||
| 14 | ПНД Ф 13.1:2:3.23-98 (ФР.1.31.2015.20483) | бутены (бутен-1, бутен-2, изобутен) | Цельностеклянный шприц на (50-100)см3 или газовая пипетка на (250-500)см3 | — | — | не более 5 часов | Шприц промывают анализируемым газом 7-10 раз, после этого производят отбор. Газовую пипетку промывают анализируемым воздухом в течение 2-3 мин. со скоростью 0,5-2,0 дм3/мин., равному 7-10 кратному объему, после этого производят отбор перекрывая одновременно оба зажима и выключают аспиратор. | ||||||||
| Этен (этилен) | |||||||||||||||
| Пропен (пропилен) | |||||||||||||||
| С1-С5 (пред.угл) | |||||||||||||||
| 15 | ФР.1.31.2004.01259 (АЮВ 0.005.169 МВИ) | Гексан | сорбционная трубка-концентратор (сорбент TENAX) | 0,05-0,1 дм3/мин | 2 мин | 2 недели в холодильни-ке | Отбирают последовательно 3 пробы в течение 20 минут. | ||||||||
| Декан | |||||||||||||||
| Акролеин (альдегид акролеиновый, проп-2-ен-1-аль) | |||||||||||||||
| Метилэтилкетон (бутан-2-он) | |||||||||||||||
| Ацетон (пропан-2-он) | |||||||||||||||
| Гексанон (4-метил-2-пентанон) | |||||||||||||||
| Циклогексанон | |||||||||||||||
| Бензол | |||||||||||||||
| Изопропилбензол (кумол) | |||||||||||||||
| О-Ксилол (1,2-диметилбензол) | |||||||||||||||
| М,П-Ксилолы (сумма) | |||||||||||||||
| Стирол (этенилбензол) | |||||||||||||||
| Толуол (метилбензол) | |||||||||||||||
| Этилбензол | |||||||||||||||
| ФР.1.31.2004.01259 (АЮВ 0.005.169 МВИ) | Бутан-1-ол | сорбционная трубка-концентратор (сорбент TENAX) | 0,05-0,1 дм3/мин | 2 мин | 2 недели в холодильни-ке | Отбирают последовательно 3 пробы в течение 20 минут. | |||||||||
| Амиловый спирт (пентанол) | |||||||||||||||
| Изобутанол (2-метилпропан-1-ол) | |||||||||||||||
| Пропиловый спирт (пропанол) | |||||||||||||||
| Изопентанол (изоамиловый спирт, 3-метил-1-бутанол) | |||||||||||||||
| изопропиловый спирт (пропан-2-ол) | |||||||||||||||
| Этанол (этиловый спирт) | |||||||||||||||
| Фенол (гидроксибензол) | |||||||||||||||
| Бутилацетат | |||||||||||||||
| Изоамилацетат | |||||||||||||||
| Винилацетат (этенилацетат) | |||||||||||||||
| Этилцеллозольв | |||||||||||||||
| Этилацетат | |||||||||||||||
| 16 | ПНД Ф 13.1:2:3.25-99 (ФР.1.31.2015.20480) | С2-С5 (непредельные) (суммарно); | Цельностеклянный шприц на (50-100)см3 или газовая пипетка на (250-500)см3 | — | — | не более 5 часов | Шприц промывают анализируемым газом 7-10 раз, после этого производят отбор. Газовую пипетку промывают анализируемым воздухом в течение 2-3 мин. со скоростью 0,5-2,0 дм3/мин., равному 7-10 кратному объему, после этого производят отбор перекрывая одновременно оба зажима и выключают аспиратор. | ||||||||
| С1-С10 (предельные) (суммарно) | |||||||||||||||
| Бензол | |||||||||||||||
| Толуол | |||||||||||||||
| Этилбензол | |||||||||||||||
| Ксилолы | |||||||||||||||
| Стирол | |||||||||||||||
| 17 | ФР.1.31.2011.11278 (М-16) | Формальдегид | Поглотительнй прибор с пористой пластинкой | 1 дм3/мин | 20 | 3 дня | Стеклянный пробоотборный зонд, носик которого заполнен стекловолокном на высоту 10 мм для устранения мешающего влияния взвешенных и смолистых веществ. Отбор проб производят одновременно из двух перпендикулярно расположенных отверстий | ||||||||
| 18 | ФР.1.31.2011.11269 (М-24) | изоционаты и ароматические амины (анилин, н-нитроанилин, толуилендиизоционат) | 2 последовательно соединенные сорбционные трубки | ПВ: 5 дм3/мин | 20 | 2 дня | Пробоотборный зонд, носик которого заполнен стекловолокном на высоту 10 мм для устранения мешающего влияния взвешенных. Отбор проб производят одновременно из двух отверстий (2 линии пробоотбора), 2 последовательно соединенные трубки присоединяют вертикально сорбентом вниз, ток газа проходит через стеклянные гранулы снизу вверх. | ||||||||
| атм. возд.: 5 дм3/мин | 60 | Отбор проб производят одновременно по 2 линиям, в каждой линии по 2 последовательно соединенные сорбционные трубки, установленные вертикально сорбентом вниз. | |||||||||||||
| РЗ: 5 дм3/мин | 15 | Отбор проб производят одновременно по 2 линиям, в каждой линии по 2 последовательно соединенные сорбционные трубки, установленные вертикально сорбентом вниз. | |||||||||||||
| 19 | ПНД Ф 13.1:2:3.59-07 (ФР.1.31.2013.16458) (М 01-05) | углеводороды алифатические предельные керосиновой фракции С12-С19 | одноразовый пробоотбоник типа «карбон» | 0,2дм3/мин | 1-3 пробы в течение 20 мин отбир. последовательно | 7 дней в холодильнике | 1-3 пробы в течение 20 минут отбирают последовательно | ||||||||
| 20 | ПНД Ф 13.1.56.-07 (ФР.1.31.2013.1643) (М-03-06) | уксусный альдегид | сорбционная трубка-концентратор (сорбент TENAX) | 0,1 дм3/мин | 20 | 3 дня в морозилке | Перед отбором пробы протягивают с помощью аспиратора газовоздушную смесь из газохода с объемным расходом 1 дм/мин в течение 5-10 мин. Затем к зонду присоединяют встык сорбционную трубку с остальными частями установки для отбора проб. Сорбционную трубку помещают в емкость со снегом или мелко колотым льдом так, чтобы ее концы оставались сухими, включают аспиратор. Интенсивное охлаждение трубки необходимо для предотвращения потерь ацетальдегида за счет проскока. | ||||||||
| пропионовый альдегид | |||||||||||||||
| маслянный альдегид | |||||||||||||||
| изомаслянный альдегид | |||||||||||||||
| 21 | ПНД Ф 13.1.54-2007 | муравьиная к-та | стеклянная трубка с сорбентом динохром Н или П | см. примечание | см. примечание | — | Ориентировочная суммарная концентрация кислот, мг/м3 | Расход при отборе проб, дм3/мин | Время отбора, мин | ||||||
| уксусная к-та | до 50 | 5 | 30 | ||||||||||||
| пропионовая к-та | от 50 до 200 вкл | 4 | 25 | ||||||||||||
| маслянная к-та | от 200 до 600 вкл | 3 | 10 | ||||||||||||
| валериановая к-та | от 600 до 2000 вкл | 2 | 5 | ||||||||||||
| капроновая к-та | |||||||||||||||
| 22 | ПНД Ф 13.1:3.68-09 (ФР.1.31.2015.19226) | Бензол | поглотительный прибор с пористой пластинкой | 0,5 дм3/мин | 20 | 3 дня в холодильнике | Не менее 3-х проб. Внимание: рекомендуемые условия отбора пробы обеспечивают достижение равновесной концентрации ароматических углеводородов в уксусной кислоте при температурах в газоходе или атмосферного воздуха от минус 20° до плюс 40°C. Срок хранения проб — 3 дня в холодильнике. При необходимости более длительного хранения отобранной пробы уксуснокислый концентрат переносят в стеклянную ампулу на 2 мл и запаивают на спиртовке. | ||||||||
| Ксилол (диметилбензол) | |||||||||||||||
| 23 | ПНД Ф13.1.8-97 (ФР.1.31.2013.16439) | бензин | одноразовый пробоотбоник типа «карбон» | (0,1-0,2) дм3/мин | Объем пробы (0,1-20) дм3 | 7 дней | После отбора проб газа пробоотборники с ВУС помещают в пробирки с притертыми пробками и доставляют в лабораторию. | ||||||||
| Концентрация вещества в газовых выбросах (мг/м3) | Объем отбираемой пробы газовых выбросов (дм3) | ||||||||||||||
| от 1 до 10 | 20 | ||||||||||||||
| Уайт-спирит | св 10 до 50 | 10 | |||||||||||||
| св 50 до 500 | 3-6 | ||||||||||||||
| Пентановая к-та (валериановая) | св 500 до 5000 | 0,3-0,6 | |||||||||||||
| св 5000 до 15000 | 0.1 | ||||||||||||||
| 24 | ПНД Ф 13.1.6-97 | Керосин | одноразовый пробоотбоник типа «карбон» | (0,1-0,3) дм3/мин | Объем пробы (0,1-20) дм3 | 7 дней | После отбора проб газа пробоотборники с ВУС помещают в пробирки с притертыми пробками и доставляют в лабораторию. | ||||||||
| Концентрация вещества в газовых выбросах (мг/м3) | Объем отбираемой пробы газовых выбросов (дм3) | ||||||||||||||
| от 1 до 10 | 20 | ||||||||||||||
| св 10 до 50 | 10 | ||||||||||||||
| св 50 до 500 | 3-6 | ||||||||||||||
| св 500 до 5000 | 0,3-0,6 | ||||||||||||||
| св 5000 до 15000 | 0.1 | ||||||||||||||
| 25 | ФР.1.31.2011.11270 (М-4) | Масло минеральное (аэрозоль масла) | обеззоленный фильтр «синяя» лента | 20 дм3/мин | 20 | 7 дней | Каждый раз отбирают масло, используемое в данном техпроцессе. Пробоотборная трубка из нержавеющей стали со сменными наконечниками. Отбирают 3-5 проб | ||||||||
| 26 | ПНД Ф 13.1.30-02 | Скипидар | одноразовый пробоотбоник типа «карбон» | (0,1-0,3) дм3/мин | Объем пробы (0,1-20) дм3 | 7 дней | После отбора проб газа пробоотборники с ВУС помещают в пробирки с притертыми пробками и доставляют в лабораторию. | ||||||||
| Концентрация вещества в газовых выбросах (мг/м3) | Объем отбираемой пробы газовых выбросов (дм3) | ||||||||||||||
| от 1 до 10 | 20 | ||||||||||||||
| св 10 до 50 | 10 | ||||||||||||||
| св 50 до 500 | 3-6 | ||||||||||||||
| св 500 до 5000 | 0,3-0,6 | ||||||||||||||
| св 5000 до 15000 | 0.1 | ||||||||||||||
| 27 | ПНД Ф 13.1.31-02 | Хром | АФА-ХП-10; АФА-ХП-20; патроны со стекловолокном | (10-20) дм3/мин | объем отобранной пробы 300 дм3 | — | — | ||||||||
| 28 | ПНД Ф 13.1:2:3.71-11 (ФР.1.31.2015.21767) | Алюминий | АФА-ВП; АФА-ХП; АФА-ХА | (10-15) дм3/мин | 20 | 1 месяц | — | ||||||||
| Барий | |||||||||||||||
| Бериллий | |||||||||||||||
| Ванадий | |||||||||||||||
| Висмут | |||||||||||||||
| Вольфрам | |||||||||||||||
| Железо | |||||||||||||||
| Галлий | |||||||||||||||
| Кадмий | |||||||||||||||
| Кобальт | |||||||||||||||
| Кремний | |||||||||||||||
| Литий | |||||||||||||||
| Магний | |||||||||||||||
| Марганец | |||||||||||||||
| Мышьяк | |||||||||||||||
| Медь | |||||||||||||||
| Молибден | |||||||||||||||
| Серебро | |||||||||||||||
| Никель | |||||||||||||||
| Олово | |||||||||||||||
| ПНД Ф 13.1:2:3.71-11 (ФР.1.31.2015.21767) | Ртуть | АФА-ВП; АФА-ХП; АФА-ХА | (10-15) дм3/мин | 20 | 1 месяц | ||||||||||
| Свинец | |||||||||||||||
| Селен | |||||||||||||||
| Сурьма | |||||||||||||||
| Титан | |||||||||||||||
| Хром | |||||||||||||||
| Цинк | |||||||||||||||
| 29 | ФР.1.31.2004.01258 (МВИ-М-34-04) | Алюминий | АФА-ВП; АФА-ХП; АФА-ХА | (15-30)дм3 | 20 | 1 месяц | — | ||||||||
| Барий | |||||||||||||||
| Бериллий | |||||||||||||||
| Ванадий | |||||||||||||||
| Висмут | |||||||||||||||
| Вольфрам | |||||||||||||||
| Железо | |||||||||||||||
| Кадмий | |||||||||||||||
| Кальций | |||||||||||||||
| Калий | |||||||||||||||
| Кобальт | |||||||||||||||
| Кремний | |||||||||||||||
| Магний | |||||||||||||||
| Марганец | |||||||||||||||
| Медь | |||||||||||||||
| Молибден | |||||||||||||||
| Мышьяк | |||||||||||||||
| Натрий | |||||||||||||||
| Никель | |||||||||||||||
| Олово | |||||||||||||||
| Ртуть | |||||||||||||||
| Селен | |||||||||||||||
| Свинец | |||||||||||||||
| Сурьма | |||||||||||||||
| Титан | |||||||||||||||
| Хром | |||||||||||||||
| Цинк | |||||||||||||||
| 30 | ПНД Ф 13.1.76-15 (ФР.1.31.2015.20718) (М 06-0-2015) | Бенз(а)пирен | фиольтры АФА-ВП-20 | 20 дм3/мин | 20 мин. | — | — |
Отбор проб воздуха рабочей зоны
| № | № НД | Название определяемой характеристики | На что отбирать | Скорость отбора | Время отбора | Срок хранения | Примечание | |||
| 1 | ФР.1.31.2001.00384 | Углерод (сажа) | АФА-ВП-20, АФА-ВП-10 | (1-40) дм³/мин | 20 | — | — | |||
| 2 | ГОСТ Р ИСО 16017-1 | Ацетонитрил | сорбционная трубка-концентратор (сорбент TENAX) | (1-10) дм³/мин | — | 8 часов ( или сорбционные трубки помещают в чистый без покрытия охлаждаемй герметичный контейнер. | — | |||
| Акрилонитрил (проп-2-енонитрил) | ||||||||||
| Гексан | ||||||||||
| Гептан | ||||||||||
| Декан | ||||||||||
| Додекан | ||||||||||
| Нонан | ||||||||||
| Октан | ||||||||||
| Углеводороды алифатические предельные бензиновой фракции С6-С12 | ||||||||||
| Ундекан | ||||||||||
| Ацетон (пропан-2-он) | ||||||||||
| Метилэтилкетон (Бутанон) | ||||||||||
| Гексанон | ||||||||||
| Диацетоновый спирт (4-гидрокси-4-метил-2-пентанон) | ||||||||||
| Циклогексанон | ||||||||||
| Бензол | ||||||||||
| Изопрпилбензол (Кумол) | ||||||||||
| Ксилол (диметилбензол) | ||||||||||
| ГОСТ Р ИСО 16017-1 | Псевдокумол (1,2,4- триметилбензол) | сорбционная трубка-концентратор (сорбент TENAX) | (1-10) дм³/мин | — | 8 часов ( или сорбционные трубки помещают в чистый без покрытия охлаждаемй герметичный контейнер. | — | ||||
| Стирол (этенилбензол) | ||||||||||
| Толуол (метилбензол) | ||||||||||
| Этилбензол | ||||||||||
| Винилхлорид (хлорэтен) | ||||||||||
| Дихлорметан | ||||||||||
| 1.1-дихлорэтилен | ||||||||||
| Трихлорэтан | ||||||||||
| Трихлорэтилен | ||||||||||
| Четыреххлористый углерод (тетрахлорметан) | ||||||||||
| Хлорбензол | ||||||||||
| Хлороформ (трихлорметан) | ||||||||||
| Бутанол | ||||||||||
| 1.2-дихлорэтан | ||||||||||
| Изобутанол | ||||||||||
| (2-метилпропан-1-ол) | ||||||||||
| Метанол (карбинол) | ||||||||||
| Амиловый спирт (пентанол) | ||||||||||
| Пропиловый спирт (прпан-1-ол) | ||||||||||
| Циклогексанол (циклогексиловый спирт) | ||||||||||
| Этанол (этиловый спирт) | ||||||||||
| Фенол (гидроксибензол) | ||||||||||
| Бутилацетат | ||||||||||
| Дибутилфталат | ||||||||||
| Диметилфталат | ||||||||||
| Демитилизофталат | ||||||||||
| Диэтилфталат | ||||||||||
| Изобутилацетат | ||||||||||
| Метилацетат | ||||||||||
| Пропилацетат | ||||||||||
| Винилацетат (Этенилацетат) | ||||||||||
| Этилакрилат | ||||||||||
| Этилацетат | ||||||||||
| 3 | ФР.1.31.2011.11272 (М-22) | Дивинил (бута-1,3-диен) | сорбционная трубка-концентратор (сорбент TENAX) | (0,05-0,2) дм³/мин | 1-3 пробы в течение 15 минут | 7 дней в холодильнике | — | |||
| Бензальдегид | ||||||||||
| Метилацетат | ||||||||||
| Изопропилацетат (1-метиэтилацетат) | ||||||||||
| н-прпилацетат | ||||||||||
| изобутилацетат | ||||||||||
| Н-Бутилацетат | ||||||||||
| Н-Амилацетат | ||||||||||
| Изопрен | ||||||||||
| Н-гексанол | ||||||||||
| ФР.1.31.2011.11272 (М-22) | 2-этилгексанол | сорбционная трубка-концентратор (сорбент TENAX) | (0,05-0,2) дм³/мин | 1-3 пробы в течение 15 минут | 7 дней в холодильнике | — | ||||
| октан-1-ол (н-октиловый спирт) | ||||||||||
| Бензиловый спирт (бензилкарбинол) | ||||||||||
| Пропионовая кислота | ||||||||||
| Пентановая к-та (валериановая) | ||||||||||
| Гексановая к-та (капроновая) | ||||||||||
| 2-этоксиэтиловый эфир уксусной к-ты | ||||||||||
| Метилцеллозольв (2-метаксиэтанол) | ||||||||||
| Изопрпилцеллозольв (2-изопропоксиэтанол) | ||||||||||
| Дифениловый эфир | ||||||||||
| Бутилцеллозольв (2-бутоксиэтанол) | ||||||||||
| Дурол (1,2,4,5-тетраметилбензол) | ||||||||||
| 1-метоксипрпопан-2-ол | ||||||||||
| 1-этоксипрпан-2-ол | ||||||||||
| 4-метилпентан-2-ол | ||||||||||
| Циклогексан | ||||||||||
| Метилбутонат | ||||||||||
| Этилбутаноат | ||||||||||
| Метилпропионат | ||||||||||
| Этилпропионат | ||||||||||
| Этиленгликоль (1,2-этандиол) | ||||||||||
| Пропиленгликоль (1,2-пропандиол) | ||||||||||
| 4 | ПНД Ф 13.1:2:3.23-98 (ФР.1.31.2015.20483) | бутены (бутен-1, бутен-2, изобутен) | шприцы | 7-10 кратная прокачка шприца | — | 5 часов | — | |||
| Этен (этилен) | ||||||||||
| Пропен (пропилен) | ||||||||||
| С1-С5 (пред.угл) | ||||||||||
| 5 | ФР.1.31.2004.01259 (АЮВ 0.005.169 МВИ) | Гексан | сорбционная трубка-концентратор (сорбент TENAX) | 0,05-0,1 дм³/мин | 2 мин | 2 недели в холодильнике | — | |||
| Декан | ||||||||||
| Акролеин (альдегид акролеиновый, проп-2-ен-1-аль) | ||||||||||
| Метилэтилкетон (бутан-2-он) | ||||||||||
| Ацетон (пропан-2-он) | ||||||||||
| Гексанон (4-метил-2-пентанон) | ||||||||||
| Циклогексанон | ||||||||||
| Бензол | ||||||||||
| Изопропилбензол (кумол) | ||||||||||
| О-Ксилол (1,2-диметилбензол) | ||||||||||
| ФР.1.31.2004.01259 (АЮВ 0.005.169 МВИ) | М,П-Ксилолы (сумма) | сорбционная трубка-концентратор (сорбент TENAX) | 0,05-0,1 дм³/мин | 2 мин | 2 недели в холодильнике | — | ||||
| Стирол (этенилбензол) | ||||||||||
| Толуол (метилбензол) | ||||||||||
| Этилбензол | ||||||||||
| Бутан-1-ол | ||||||||||
| Амиловый спирт (пентанол) | ||||||||||
| Изобутанол (2-метилпропан-1-ол) | ||||||||||
| Пропиловый спирт (пропанол) | ||||||||||
| Изопентанол (изоамиловый спирт, 3-метил-1-бутанол) | ||||||||||
| изопропиловый спирт (пропан-2-ол) | ||||||||||
| Этанол (этиловый спирт) | ||||||||||
| Фенол (гидроксибензол) | ||||||||||
| Бутилацетат | ||||||||||
| Изоамилацетат | ||||||||||
| Винилацетат (этенилацетат) | ||||||||||
| Этилцеллозольв | ||||||||||
| Этилацетат | ||||||||||
| 6 | ПНД Ф 13.1:2:3.25-99 (ФР.1.31.2015.20480) | С2-С5 (непредельные) (суммарно); | шприцы | 7-10 кратная прокачка шприца | — | 5 часов | — | |||
| С1-С10 (предельные) (суммарно) | ||||||||||
| Бензол | ||||||||||
| Толуол | ||||||||||
| Этилбензол | ||||||||||
| Ксилолы | ||||||||||
| Стирол | ||||||||||
| 7 | ФР.1.31.2011.11269 (М-24) | изоционаты и ароматические амины (анилин, н-нитроанилин, толуилендиизоционат) | 2 последовательно соединенные сорбционные трубки СТ 412 | 5 дм³/мин | 15 | 2 дня в холодильнике | 2 линии пробоотбора, в каждой по 2 последовательно соединенные сорбционные трубки, установленные вертикально сорбентом вниз. | |||
| 8 | ПНД Ф 13.1:2:3.59-07 (ФР.1.31.2013.16458) (М 01-05) | углеводороды алифатические предельные керосиновой фракции С12-С19 | одноразовый пробоотбоник типа «карбон» | 0,2-0,3 дм³/мин | 15 | 7 дней в хоодильнике | в период 15 мин отбирают 3 последовательные пробы | |||
| 9 | МУК 4.1.1957-05 | винилхлорид | одновременно на 2 сорбционные трубки (сорбент Chromosorb 104) | 0,2 дм³/мин | 10 | 3 дня в холодильнике | Каждая проба воздуха одновременно отбирается на две сорбционные трубки с помощью 2 пробоотборников. Воздух прокачивают через узкое отверстие сорбционной трубки, охлаждаемой холодильником со льдом до 0 °С. |
|||
| ацетальдегид | ||||||||||
| 10 | МУК 4.1.1143-02 | Тиметоксам | фильтр «синяя лента» | 4 дм³/мин | 5 | в морозильной камере при температуре -12 °С в течение 30 дней, в холодильнике при 4 — 6 °С — в течение 7 дней. | — | |||
| 11 | ФР.1.31.2004.01258 (МВИ-М-34-04) | Алюминий | АФА-ВП; АФА-ХП; АФА-ХА | 10 дм³/мин | 15 | 1 месяц | — | |||
| Барий | 20 дм³/мин | 30 | ||||||||
| Бериллий | 20 дм³/мин | 30 | ||||||||
| Ванадий | 20 дм³/мин | 30 | ||||||||
| Висмут | 20 дм³/мин | 30 | ||||||||
| Вольфрам | 20 дм³/мин | 15 | ||||||||
| Железо | 20 дм³/мин | 30 | ||||||||
| Кадмий | 20 дм³/мин | 30 | ||||||||
| Кальций | 20 дм³/мин | 15 | ||||||||
| Калий | 20 дм³/мин | 20 | ||||||||
| Кобальт | 10 дм³/мин | 15 | ||||||||
| Кремний | 20 дм³/мин | 15 | ||||||||
| Магний | 10 дм³/мин | 15 | ||||||||
| Марганец | 10 дм³/мин | 20 | ||||||||
| Медь | 10 дм³/мин | 15 | ||||||||
| Молибден | 20 дм³/мин | 30 | ||||||||
| Мышьяк | 20 дм³/мин | 30 | ||||||||
| Натрий | 20 дм³/мин | 20 | ||||||||
| Никель | 10 дм³/мин | 20 | ||||||||
| Олово | 20 дм³/мин | 30 | ||||||||
| Ртуть | 20 дм³/мин | 20 | ||||||||
| Селен | 20 дм³/мин | 30 | ||||||||
| Свинец | 20 дм³/мин | 30 | ||||||||
| Сурьма | 20 дм³/мин | 15 | ||||||||
| Титан | 10 дм³/мин | 15 | ||||||||
| Хром | 20 дм³/мин | 30 | ||||||||
| Цинк | 10 дм³/мин | 20 |
Отбор проб воздуха замкнутых помещений
| № | № НД | Название определяемой характеристики | На что отбирать | Скорость отбора | Время отбора | Срок хранения | Примечание | ||
| 1 | ГОСТ Р ИСО 16000-3 | Бензальдегид (альдегид бензойный) | картриджей с нанесенным на силикагель ДНФГ | 1 дм³/мин | 5-60 | 30 дней в холодильнике, без холодильника не более 2 дней | — | ||
| Альдегтд диметилбензойный (2.5-диметилбензальдегид) | |||||||||
| Альдегтд валериановый (валеральдегид)) | |||||||||
| альдегид изовалериановый | |||||||||
| гексаналь (альдегид капроновый) | |||||||||
| альдегид кротоновый | |||||||||
| альдегид пропионовый (пропиональ) | |||||||||
| о-толуоловый альдегид | |||||||||
| м-толуоловый альдегид | |||||||||
| п-толуоловый альдегид | |||||||||
| ацетальдегид (альдегид уксусный) | |||||||||
| альдегид маслянный (бутаналь) | |||||||||
| формальдегид (метаналь) | |||||||||
| ацетон (пропан-2-он) | |||||||||
| 2 | МУК 4.1.1957-05 | винилхлорид | одна проба состоит из 2 сорбционнх трубок (сорбент Chromosorb 104) | 0,2 дм³/мин | 10 | 3 дня в холодильнике | Воздух прокачивают со скоростью 0,2 дм3/мин в течение 10 мин через узкое отверстие сорбционной трубки, охлаждаемой холодильником со льдом до 0 °С. | ||
| ацетальдегид | |||||||||
| 3 | ГОСТ Р ИСО 16000-14 | ПХБ (инд.) | сорбент с нанесенным изотопом С13 | — | — | — | — | ||
| 4 | МУК 4.1.1044а-01 | Диметиламин | одновременно на 2 сорбционные трубки (сорбент Chromosorb 106 или 103) | 0,2 дм³/мин | 10 | 3 дня | Воздух аспирируют через предварительно охлажденное до 0°С отверсие сорбционной трубки. | ||
| Диэтиламины | |||||||||
| Пропиламины | |||||||||
| Триэтиламины | |||||||||
| Этиламины | |||||||||
| Диметилформамид | |||||||||
| 5 | ГОСТ Р ИСО 16017-1 | Ацетонитрил | сорбционная трубка-концентратор (сорбент TENAX) | (1-10) дм³/мин | — | 8 часов ( или сорбционные трубки помещают в чистый без покрытия охлаждаемй герметичный контейнер. | — | ||
| Акрилонитрил (проп-2-енонитрил) | |||||||||
| Гексан | |||||||||
| Гептан | |||||||||
| Декан | |||||||||
| Додекан | |||||||||
| Нонан | |||||||||
| Октан | |||||||||
| Углеводороды алифатические предельные бензиновой фракции С6-С12 | |||||||||
| Ундекан | |||||||||
| Ацетон (пропан-2-он) | |||||||||
| Метилэтилкетон (Бутанон) | |||||||||
| Гексанон | |||||||||
| Диацетоновый спирт (4-гидрокси-4-метил-2-пентанон) | |||||||||
| Циклогексанон | |||||||||
| Бензол | |||||||||
| Изопрпилбензол (Кумол) | |||||||||
| Ксилол (диметилбензол) | |||||||||
| Псевдокумол (1,2,4- триметилбензол) | |||||||||
| Стирол (этенилбензол) | |||||||||
| Толуол (метилбензол) | |||||||||
| Этилбензол | |||||||||
| Винилхлорид (хлорэтен) | |||||||||
| Дихлорметан | |||||||||
| 1.1-дихлорэтилен | |||||||||
| Трихлорэтан | |||||||||
| Трихлорэтилен | |||||||||
| ГОСТ Р ИСО 16017-1 | Четыреххлористый углерод (тетрахлорметан) | сорбционная трубка-концентратор (сорбент TENAX) | (1-10) дм³/мин | — | 8 часов ( или сорбционные трубки помещают в чистый без покрытия охлаждаемй герметичный контейнер. | — | |||
| Хлорбензол | |||||||||
| Хлороформ (трихлорметан) | |||||||||
| Бутанол | |||||||||
| 1.2-дихлорэтан | |||||||||
| Изобутанол | |||||||||
| (2-метилпропан-1-ол) | |||||||||
| Метанол (карбинол) | |||||||||
| Амиловый спирт (пентанол) | |||||||||
| Пропиловый спирт (прпан-1-ол) | |||||||||
| Циклогексанол (циклогексиловый спирт) | |||||||||
| Этанол (этиловый спирт) | |||||||||
| Фенол (гидроксибензол) | |||||||||
| Бутилацетат | |||||||||
| Дибутилфталат | |||||||||
| Диметилфталат | |||||||||
| Демитилизофталат | |||||||||
| Диэтилфталат | |||||||||
| Изобутилацетат | |||||||||
| Метилацетат | |||||||||
| Пропилацетат | |||||||||
| Винилацетат (Этенилацетат) | |||||||||
| Этилакрилат | |||||||||
| Этилацетат | |||||||||
| 6 | ПНД Ф 13.1:2:3.23-98 (ФР.1.31.2015.20483) | бутены (бутен-1, бутен-2, изобутен) | шприцы | 7-10 кратная прокачка шприца | — | 5 часов | — | ||
| Этен (этилен) | |||||||||
| Пропен (пропилен) | |||||||||
| С1-С5 (пред.угл) | |||||||||
| 7 | ГОСТ 17.2.4-05 | взвешенные вещва (пыль) | фильтр АФА-ВП | 75 дм³/мин | По окончании отбора регистрируют общий объем пропущенного воздуха. | — | — | ||
| 8 | ГОСТ ISO 16000-6 | 1,2,3-триметилбензол | сорбционная трубка-концентратор (сорбент TENAX) | от 50 до 200 мл³/мин | от 25 до 100 мин | 4-недели с момента отбора | — | ||
| 1,2,4-триметилбензол | |||||||||
| бензол | |||||||||
| Этилбензол | |||||||||
| изопропилбензол | |||||||||
| м-, п-Ксилолы (сумма) | |||||||||
| о-ксилол | |||||||||
| Стирол | |||||||||
| Толуол (метилбензол) | |||||||||
| 1-бутанол | |||||||||
| 1-пентанол | |||||||||
| 1-пропанол | |||||||||
| 2-метил-1-пропанол(изобутанол) | |||||||||
| циклогексанол | |||||||||
| Фенол (гидроксибензол) | |||||||||
| ацетальдегид | |||||||||
| 2-бутанон(метилэтилкетон) | |||||||||
| ацетон | |||||||||
| циклогексанон | |||||||||
| 1,1,1-трихлорэтан | |||||||||
| 1,1,2-трихлорэтан | |||||||||
| 1,2-дихлорэтан | |||||||||
| Четыреххлористый углерод (тетрахлорметан) | |||||||||
| Хлорбензол | |||||||||
| Дихлорметан | |||||||||
| трихлорметан | |||||||||
| тетрахлорэтен | |||||||||
| трихлорэтен | |||||||||
| Бутилацетат | |||||||||
| Этилацетат | |||||||||
| Этилакрилат | |||||||||
| изобутилакрилат | |||||||||
| метиакрилат | |||||||||
| метилметакрилат | |||||||||
| Пропилацетат | |||||||||
| Винилацетат (Этенилацетат) | |||||||||
| Дибутилфталат | |||||||||
| Диметилфталат | |||||||||
| гексан | |||||||||
| ГОСТ ISO 16000-6 | гептан | сорбционная трубка-концентратор (сорбент TENAX) | от 50 до 200 мл³/мин | от 25 до 100 мин | 4-недели с момента отбора | — | |||
| октан | |||||||||
| нонан | |||||||||
| декан | |||||||||
| ундекан | |||||||||
| додекан | |||||||||
| сумма СЛОС (SVOS) | |||||||||
| cумма ВЛОС(VVOC) | |||||||||
| ОЛОС (TVOC)в толуоловом эквиваленте |
РУКОВОДСТВО
ПО МЕТОДАМ ОПРЕДЕЛЕНИЯ ВРЕДНЫХ ВЕЩЕСТВ
В АТМОСФЕРНОМ ВОЗДУХЕ
РЕФЕРАТ
Санитарная охрана атмосферного воздуха приобретает особо важное значение в связи с широким развитием промышленности и автотранспорта в нашей стране. Органы санитарного контроля изучают состояние воздушной среды населенных мест, устанавливая предельно допустимые концентрации вредных веществ в воздухе. Для успешного проведения подобных исследований необходимо располагать объективными и надежными методами определения вредных веществ в атмосферном воздухе. Чем точнее полученные данные, тем эффективнее будут проведены оздоровительные мероприятия по охране атмосферного воздуха от загрязнений.
Настоящая книга является пособием по методам определения вредных веществ в атмосферном воздухе и состоит из 15 глав. В главе I изложены общие вопросы по определению атмосферных загрязнений: даны указания по отбору проб, максимально разовых и среднесуточных, подробно описаны способы отбора проб воздуха, приборы и аппаратура, применяемые для этой цели, способы приготовления стандартных растворов, формулы расчета анализа, приведения объема воздуха к нормальным условиям и т.д.
В 15 главах руководства описаны методы определения токсических веществ, распределенных по классам соединений, например соединения серы, соединения азота, металлы, аминосоединения и др.
В книге описаны методы определения отдельных токсических веществ в атмосферном воздухе. Методы составлены по единой схеме, перед изложением каждого метода приведены физико-химические и токсические свойства определяемого соединения, а также предельно допустимые концентрации — максимальные разовые и среднесуточные. Для ряда токсических веществ дано несколько методов, что расширяет возможности определения этих веществ при различных условиях.
Приведенные в руководстве методы позволяют в первую очередь определять вещества, на которые установлены предельно допустимые концентрации. Кроме того, приводится описание методов определения веществ, вновь синтезируемых и внедряемых в промышленность и являющихся источником загрязнения атмосферного воздуха.
Настоящее руководство рассчитано на химиков, работающих в санитарно-эпидемиологических станциях, лабораториях гидрометеорологической службы, а также в ведомственных лабораториях.
ПРЕДИСЛОВИЕ
Охрана внешней среды, в том числе атмосферного воздуха, в нашей стране поставлена на уровень важнейших государственных задач.
Решения XXIV съезда КПСС по усилению охраны природы в стране, Основы законодательства Союза ССР и союзных республик о здравоохранении (утверждены VII сессией Верховного Совета СССР 19 декабря 1969 г.), Постановление «О мерах по дальнейшему улучшению охраны природы и рациональному использованию природных ресурсов» (принято 20 сентября 1972 г.) целеустремленно направлены на бережное отношение к природе, в частности, на охрану чистоты атмосферного воздуха как важнейшего фактора, обеспечивающего состояние здоровья населения.
Указанные постановления партии и правительства обязывают руководителей министерств, ведомств, предприятий, учреждений, проектных и строительных организаций предусматривать в перспективных и годовых планах проведение природооздоровительных мероприятий по предупреждению загрязнения атмосферного воздуха, водоемов, подземных вод и почвы и претворять их в жизнь.
Вместе с тем рост промышленности и транспорта все еще неизбежно ведет к загрязнению атмосферы городов и других населенных пунктов. Основными источниками загрязнения воздушного бассейна вредными веществами являются ТЭЦ, котельные, транспорт, различные двигатели внутреннего сгорания, а также предприятия металлургической и химической промышленности. Следует отметить, что в связи с химизацией народного хозяйства значительно увеличился выброс в атмосферу различных соединений сложного состава, ранее не известных и не безопасных для здоровья населения.
В успешной борьбе за чистоту атмосферного воздуха существенное место занимают санитарный надзор и дальнейшие, вытекающие из обстановки гигиенические мероприятия.
Систематический санитарно-лабораторный контроль в стране осуществляют санитарно-эпидемиологические станции, лаборатории гидрометеорологической службы и в соответствии с Постановлением ЦК КПСС и Совета Министров СССР N 517 от 1968 г. ведомственные и производственные лаборатории.
Санитарный надзор по охране атмосферного воздуха является эффективным в нашей стране благодаря разработанным критериям оценки вредности различных веществ, базирующимся на основе материалистического учения И.П. Павлова. Принципы гигиенического нормирования вредных веществ в атмосферном воздухе основаны на фундаментальных исследованиях советских гигиенистов.
Гигиенические нормативы лежат в основе санитарного законодательства и позволяют получать объективные данные о степени и опасности загрязнения воздушной среды в населенных местах. В этих условиях, естественно, трудно переоценить значение объективных, надежных методов лабораторного контроля воздушной среды.
Чем точнее получены данные, тем эффективнее будут оздоровительные мероприятия по охране атмосферного воздуха от загрязнения. Следует подчеркнуть особую сложность разработки методических решений в области охраны атмосферного воздуха. С одной стороны, необходимо разработать методы, позволяющие определить микроколичества веществ, с другой — следует учитывать, что в атмосферном воздухе всегда одновременно присутствуют группы и целые комплексы различных веществ, осложняющие аналитическую работу.
Настоящая книга является фундаментальным пособием по методам определения вредных веществ в атмосферном воздухе.
Книга рассчитана главным образом на химиков, работающих в санитарно-эпидемиологических станциях, а также на работников различных ведомственных учреждений, выполняющих анализы воздушной среды.
В основу книги положен большой многолетний опыт работы авторов в области санитарной химии в Московском научно-исследовательском институте гигиены имени Ф.Ф. Эрисмана и в других научно-исследовательских институтах гигиенического профиля, также использован литературный материал.
В книге дается краткая характеристика физико-химических свойств групп веществ, приводится их токсичность, описаны наиболее точные и одновременно общедоступные методы определения этих веществ в атмосферном воздухе.
Изложение книги начинается с основ лабораторной практики, что весьма необходимо, особенно начинающим химикам.
Разработаны рекомендации по отбору максимально разовых и среднесуточных проб воздуха, имеющих принципиальное значение при гигиенической оценке воздушной среды и атмосферных загрязнений.
Подробно изложены основы современных методических исследований с учетом использования новой лабораторной техники (фотометрии, хроматографии и др.). Одновременно даны вполне доступные методы исследования для небольших лабораторий.
Директор
Московского научно-исследовательского
института гигиены имени Ф.Ф. Эрисмана
профессор
член-корреспондент АМН СССР
А.П.ШИЦКОВА
ВВЕДЕНИЕ
Советская медицина стоит на позициях предупреждения и своевременного устранения различных факторов внешней среды, оказывающих вредное влияние на здоровье человека.
В решениях XXIV съезда КПСС, в Основах законодательства Союза ССР и союзных республик, а также в Постановлении Верховного Совета СССР «О мерах по дальнейшему улучшению охраны природы и рациональному использованию природных ресурсов» задачи защиты чистоты атмосферного воздуха и предохранения его от загрязнения признаны общенародными, общегосударственными, от правильного и своевременного решения которых во многом зависит состояние здоровья населения нашей страны [11, 22, 24, 26].
Для защиты воздушного бассейна от загрязнения промышленными выбросами устанавливают мощные очистительные сооружения, газо-пылевые устройства, проводят строительство поясов зеленых насаждений вокруг городов и промышленных центров, а также проводят другие санитарно-гигиенические мероприятия.
К сожалению, отдельные очистные сооружения работают пока еще недостаточно эффективно, иногда имеют место непредусмотренные выбросы, аварийные случаи, что влечет за собой загрязнение атмосферного воздуха населенных мест. Это свидетельствует о техническом несовершенстве отдельных очистительных сооружений.
Особенно сильно загрязняет воздушный бассейн автомобильный транспорт, выхлопные газы которого с содержащимися в них токсическими соединениями находятся в прямой зависимости от режима работы двигателей.
Дальнейшее развитие химической, нефтехимической промышленности, а также черной и цветной металлургии и увеличивающееся использование двигателей внутреннего сгорания связано с повышенным количеством выбрасываемых веществ в атмосферный воздух. При этом не только растет число разнообразных химических веществ, выбрасываемых в атмосферный воздух, но и усложняется состав выбросов. В свою очередь наличие токсических веществ в атмосферном воздухе является источником как дискомфорта, так и прямой угрозы здоровью населения.
Токсические вещества имеют широкий диапазон вредного воздействия. Так, в одних случаях они вызывают токсикозы, в других создают неблагоприятные условия для произрастания и формирования растений, вызывают коррозию строительных материалов и др.
Влияние вредных атмосферных загрязнений на здоровье человека имеет свою специфику, с которой следует считаться. Дело в том, что в производственных помещениях, воздух которых загрязнен токсическими соединениями, как правило, работают взрослые. Пребывание их в помещениях, атмосферный воздух которых загрязнен токсическими соединениями, ограничивается только рабочим временем. Другое дело, когда речь идет о загрязнении атмосферного воздуха населенного пункта. В этом случае он воздействует непрерывно и днем, и ночью, в течение всего времени загрязнения воздушного бассейна и, что самое главное, его воздействию подвергаются люди всех возрастов — от самого раннего до старческого, которые особенно чувствительны к токсическим веществам.
Все это и заставляет работников здравоохранения изучать и решать вопросы, связанные с охраной атмосферного воздуха. Вполне понятно, что решить эту проблему можно лишь совместными усилиями технологов, гигиенистов, химиков, физиологов, физиков, токсикологов и специалистов других профессий.
Роль химиков в этом велика и ответственна, они определяют, какими веществами загрязнен воздух и в каком количестве. От правильного заключения будет зависеть и направленность всех проводимых оздоровительных мероприятий.
Практически контроль за состоянием воздушного бассейна в нашей стране осуществляют органы здравоохранения. В их ведении и подчинении находятся специальные лаборатории Министерства здравоохранения СССР. Кроме того, работы по исследованию атмосферного воздуха проводятся и в лабораториях ведомственных учреждений. Большую работу по исследованию воздуха ведут гидрометеорологические службы страны; проводятся крупные научно-исследовательские, гигиенические работы, связанные с анализом атмосферного воздуха.
Во исполнение Постановления ЦК КПСС и Совета Министров СССР от 5 июля 1968 г. N 517 «О мерах по дальнейшему улучшению здравоохранения и развития медицинской науки в стране» на промышленных предприятиях должна быть организована еще более широкая сеть лабораторий по осуществлению постоянного контроля за соблюдением санитарно-гигиенических нормативов, в частности за состоянием атмосферного воздуха.
Естественно, что для успешного проведения таких работ необходимо располагать методами исследования атмосферного воздуха. В данном руководстве изложены методы определения большого числа токсических веществ в атмосферном воздухе. Почти по каждому веществу представлен ряд методов, используемых в зависимости от условий.
В руководстве в основном изложены методы, используемые при исследовании атмосферного воздуха, однако отдельные высокочувствительные и проверенные на практике методы заимствованы из области исследования воздуха производственных помещений.
В настоящее время в практике исследования атмосферных загрязнений широко используются фотометрические методы, реже — хроматографические, полярографические и другие виды анализа.
Сущность отдельных видов анализа в руководстве не освещена потому, что более подробное ее описание приводится в специальной литературе.
В данном руководстве в первую очередь описаны те токсические соединения, на которые утверждены предельно допустимые концентрации (ПДК) в воздухе населенных мест.
В общей части руководства изложены некоторые основные положения, связанные с определением малых количеств токсических веществ. В специальном разделе даются методы отбора проб и определения токсических веществ в атмосферном воздухе. Методы определения отдельных токсических веществ описаны по единой схеме.
Перед изложением методов определения того или иного соединения дана краткая информация о физических и токсикологических свойствах, приведены предельно допустимые, максимально разовые и среднесуточные концентрации.
Далее описаны принципы метода, указана чувствительность в определяемом объеме или в 1 мл пробы. Также указано влияние некоторых примесей, проверенных авторами методов. Приведен список необходимых реактивов, описан отбор проб, изложен ход анализа.
Возможно, что некоторые вопросы остались неосвещенными, поэтому авторы будут благодарны за критические замечания и дополнения по данному руководству.
Глава I
ОБЩИЕ ВОПРОСЫ ПО ОПРЕДЕЛЕНИЮ АТМОСФЕРНЫХ ЗАГРЯЗНЕНИЙ
Атмосферный воздух представляет собой смесь газов: азота, кислорода, углекислого газа, аргона, неона и др. Состав атмосферного воздуха до высоты 85 — 100 км практически одинаков, в составе околоземных слоев сухого атмосферного воздуха присутствуют следующие компоненты (табл. 1).
Таблица 1
СОСТАВ СУХОГО АТМОСФЕРНОГО ВОЗДУХА [3]
|
Компоненты |
Процент по весу |
Компоненты |
Процент по весу |
|
Азот |
75,50 |
Гелий |
0,00007 |
|
Кислород |
23,12 |
Водород |
0,00005 |
|
Аргон |
1,285 |
Криптон |
0,0000025 |
|
Углекислый газ |
0,046 |
Радон |
6 · 10-18 |
|
Неон |
0,001 |
Содержание озона у поверхности земли находится в пределах 3 · 10-6 по объему. Концентрация его увеличивается с высотой и достигает максимума на уровне 25 — 30 км.
В воздухе содержится 0 — 4% водяных паров по объему.
В связи с ростом металлургической, химической и других видов промышленности атмосферный воздух загрязняется многими токсическими веществами.
В основном источником загрязнения воздушного бассейна являются промышленные предприятия, размещенные без соблюдения санитарно-гигиенических норм и правил или эксплуатирующиеся с их нарушением.
Количество токсических веществ, поступающих в атмосферный воздух от действующих предприятий, зависит от ряда обстоятельств, в том числе от объема и количества выбросов, условий поступлений их в атмосферу. Концентрация токсических веществ меняется в зависимости от высоты труб, систематичности и случайности выбросов, от удаления точек, где проводят исследование, от источника загрязнения, ландшафта местности, направления и силы ветра, градиента температуры, влажности и др.
Атмосферный воздух загрязняется также топочными газами котельных и выхлопными газами автотранспорта.
В атмосферном воздухе под действием ионизирующих излучений происходят фотохимические процессы с образованием окислов азота, озона и т.д. Озон образуется при облучении воздуха ультрафиолетовой радиацией с длиной волны 253 нм, а окислы азота — до 100 нм. Поверхности земли достигает радиация лишь с длиной волны около 290 нм.
В условиях загрязнения атмосферного воздуха промышленными выбросами, выхлопными газами автотранспорта фотохимические реакции проходят под действием обычной солнечной радиации. При этом возможно превращение окиси азота в двуокись, накопление озона в атмосфере и др. При взаимодействии углеводородов с озоном или атомарным кислородом образуются свободные пероксильные, высокореактивные вещества, способные вступать в реакцию с окислами азота и другими соединениями и образовывать сложный комплекс веществ, обладающих окислительными свойствами, — оксиданты.
При особых метеорологических условиях, способствующих накоплению оксидантов, в атмосферном воздухе может образовываться фотохимический смог, сопровождающийся уменьшением прозрачности (снижением дальности видимости), появлением неприятных запахов, раздражением слизистых оболочек глаз, носа, горла, увяданием растительности и др. С целью обнаружения токсических веществ, находящихся в виде газов, паров, аэрозолей, пыли, проводятся гигиенические исследования атмосферных загрязнений. Обычно воздух загрязнен токсическими веществами, находящимися в весьма сложном сочетании, что в ряде случаев затрудняет проведение анализа.
Санитарный контроль за чистотой атмосферного воздуха осуществляется путем отбора проб с анализом этих проб.
Исследуя атмосферный воздух как с научной, так и с практической целью, приходится выполнять анализы проб, в которых содержатся весьма малые количества веществ. Это вызвано высокой степенью разбавления выбросов в атмосфере и низкими ПДК для воздуха населенных мест [25].
Это указывает на специфические трудности, возникающие при проведении анализа атмосферных загрязнений, и налагает особую ответственность на химиков-аналитиков при выборе наиболее чувствительных и по возможности избирательных методов исследования.
Исследования атмосферного воздуха связаны с определением микрограммовых количеств веществ, и понятно, что при анализе воздушной среды должны применяться высокочувствительные методы, а сам анализ должен быть выполнен в строгом соответствии с правилами, предъявляемыми к аналитической химии малых концентраций.
При анализах воздуха высокочувствительными методами необходимо учитывать, что если определяемая величина оказывается близкой к чувствительности метода, то ошибка определения может быть весьма ощутимой. Во избежание этого следует, например, применяя колориметрические методы, использовать по возможности калибровочный график или сравнивать интенсивность окраски со шкалой в средней части графика или шкалы [19, 20].
При определении микроколичеств следует учитывать также наличие сопутствующих веществ, иногда превышающих искомое вещество, а также фон, который образуется постоянными или случайными загрязнениями, попадающими из реактивов, посуды и т.п.
Применяемая посуда должна быть тщательно промыта и пропарена. Приспособление для пропаривания показано на рис. 1.
Рис. 1. Приспособление для пропаривания
При проведении анализов проб атмосферного воздуха необходимо устранять возможные потери, происходящие за счет улетучивания, адсорбции, ионного обмена; частицы золы могут быть унесены конвенционными потоками и др. Следует обращать внимание и на потери, которые могут быть при применении некачественной фарфоровой посуды (трещины, вплавление в стенки сосуда и др.). Во избежание этого необходимо тщательно соблюдать режим нагревания, не допускать разбрызгивания и соблюдать другие предосторожности. С целью, например, устранения потерь искомого вещества при фильтровании можно пользоваться малой массой фильтровальной бумаги. Для этого делают соскоб с фильтра и получают волокна. Фильтрование ведут в небольшой воронке с оттянутым концом (рис. 2), в который вкладывают тампон из волокон фильтра длиной 0,2 — 0,3 см (вес около 0,05 г). Если возможно по ходу анализа, волокна в воронке предварительно промывают разбавленным раствором кислоты.
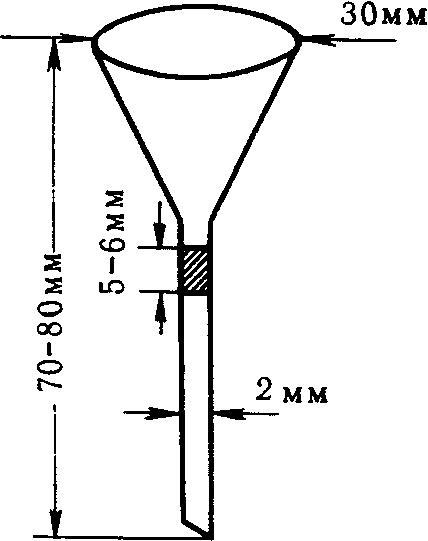
Рис. 2. Воронка для фильтрования
С целью нивелирования погрешности анализа обязательно подготавливают контрольное определение, проведенное в идентичных с анализом пробы условиях. Повышение чувствительности определения достигается различными приемами. Применяется способ концентрирования исследуемого вещества в процессе отбора проб, что осуществляется увеличением скорости протягивания воздуха в единицу времени или увеличением времени отбора пробы. При применении жидких поглотительных сред указанный прием возможен лишь при получении в процессе поглощения нелетучего продукта.
Достигается это и в процессе анализа концентрированием пробы, упариванием, отгонкой, экстрагированием, хроматографированием, уменьшением растворимости осадка; применяются приборы с большей разрешающей способностью, используются специальные приспособления для кювет и др. [7, 9].
Некоторые методы достаточно чувствительны и не требуют накопления веществ, что позволяет проводить определение в малом объеме (до 1 л). При этом отбирают пробы воздуха в газовые пипетки.
Определение токсических веществ в атмосферном воздухе складывается из двух основных разделов — отбора проб и последующего анализа. Оба раздела имеют самостоятельное значение, дополняют друг друга, и качество отбора проб влияет на результат определения. Критерием оценки загрязнения являются данные, полученные в результате отбора и анализа максимальных разовых и среднесуточных концентраций вредных веществ в атмосферном воздухе [1, 6].
ОТБОР ПРОБ ВОЗДУХА [14]
Постоянный контроль за состоянием атмосферного воздуха осуществляется путем отбора максимальных разовых и среднесуточных проб воздуха в точках, расположенных в заранее определяемых местах. С целью правильного размещения точек отбора проб необходимо располагать планом исследуемого района с нанесением на этот план основных ориентиров. Следует иметь данные, характеризующие то или иное предприятие как источник загрязнения атмосферного воздуха. Это поможет установить время отбора проб воздуха в течение суток или сезона, а также определить расстояния, на которых необходимо вести отбор проб воздуха.
При расстановке точек отбора проб учитывают метеорологические и рельефные особенности размещения производств, сложившуюся ситуацию по взаиморазмещению объектов и жилых районов, а также другие специфические особенности.
Одновременно с отбором проб на всех точках производят наблюдения за скоростью и направлением ветра, отмечают атмосферное давление и температуру.
Отбор максимальных разовых проб. При изучении загрязнений атмосферного воздуха проводят отбор проб в зонах максимального загрязнения, непосредственно в факеле выброса. Отбор проб может быть проведен последовательно в каждой зоне обязательно с захватом факела.
Лучшим вариантом отбора максимальных разовых проб следует считать одновременный отбор во всех обследуемых зонах, причем в 2 — 4 точках, расположенных поперек в каждой зоне факела. Расстояние крайних точек относительно осевой обычно не превышает 5% расстояния от источника выброса до зоны исследования (например, в зоне 2000 м 2 точки должны быть расставлены на расстоянии не более 100 м от точки, находящейся на оси). При таком отборе проб учитывают максимальные показатели, боковые точки лишь подтверждают правильность расстановки точек.
Отбор проб под факелом осуществляется, как правило, в течение 20 мин. и не превышает 30 мин. В каждой точке отбирают не менее 25 проб в течение ряда дней на уровне 1,5 м от земли (в зоне дыхания). При большом разбросе полученных данных эта величина может оказаться недостаточной.
В отдельных случаях отбирают разовые пробы в стационарах или подвижных точках без учета факела. В этих случаях отбор проб лучше проводить то в утренние, то в вечерние часы суток, в часы максимального загрязнения воздуха. Отбирают пробы на уровне 1,5 м от земли (в зоне дыхания), в местах, где нет пыли, например на газоне, твердом грунте и т.п. Отбор проб в подвижных и стационарных точках обычно не превышает 30 мин.
Отбор проб проводят аспирированием исследуемого воздуха через поглотительный прибор, в который помещен тот или иной сорбент, накапливают токсическое вещество до необходимых пределов. В отдельных случаях отбор проб проводят за короткий промежуток времени, например в газовые пипетки. При этом с целью получения средних данных по концентрации токсических веществ за 20 — 30 мин. можно отбирать последовательно 5 — 6 проб с одинаковыми интервалами.
Отбор среднесуточных проб. Среднесуточные пробы отбирают на стационарных и подвижных точках, в зоне дыхания человека на высоте 1,5 м от земли, на открытых площадках в удалении от строений. Выбирают точки отбора там, где нет пыли. В стационарных точках отбор проб проводят периодически, не меняя их расположения. В подвижных точках отбор проб проводят также в течение суток, но, в зависимости от изменяющихся условий, перемещают точки отбора.
При отборе проб в стационарных точках не учитывают направление ветра, и, если пункт наблюдения выходит из факела, отбор проб продолжают.
Обычно для отбора проб выбирают период наибольшего загрязнения атмосферного воздуха по условиям выброса или метеорологическим данным. Желательно размещать как можно больше точек отбора, но если такой возможности нет, то при отборе проб в передвижных точках следует в разные дни выезда менять их расположение.
Рекомендуется отбирать среднесуточные пробы в количестве не менее 10 — 15 в сезон. Пробы отбирают в течение суток, причем допускается несколько вариантов исследований:
1) исследуемый воздух протягивают непрерывно в течение суток через один и тот же поглотительный прибор, наполненный поглотительным раствором, через фильтр, помещенный в патрон, или твердый сорбент;
2) исследуемый воздух протягивают через один и тот же поглотительный прибор, наполненный поглотительным раствором, через фильтр или твердый сорбент, с перерывами в определенные промежутки времени, через 2 или 4 ч (без смены поглотительного прибора, патрона). Отбирают пробы в течение суток 6 или 12 раз по 20 — 30 мин.;
3) в случае необходимости нужно проследить за динамикой загрязнения воздуха района, в различные часы суток можно использовать способ отбора проб в разные поглотительные приборы, т.е. провести отбор проб, например, 6, 12 или 24 раза через одинаковые промежутки времени, при этом воздух протягивают в течение 20 — 30 мин. и каждую пробу анализируют отдельно. При таком способе удается установить периоды максимального и минимального загрязнения.
При отборе проб воздуха необходимо учитывать состояние определяемого вещества в воздухе (газ, пар, аэрозоли).
В зависимости от этого отбор проб осуществляют в поглотительные приборы, содержащие поглотительные среды, или на фильтрующие материалы, помещенные в патроне.
Для поглощения токсических веществ из воздуха применяют аспирационные способы или способы отбора проб в сосуды ограниченной емкости.
Аспирационные способы отбора проб. Отбор проб атмосферного воздуха проводят в населенных пунктах и в полевых условиях, вдали от населенных мест и от производства. В зависимости от условий применяют различные аспирационные устройства. Используют аспираторы, работающие от электросети или аккумуляторов, применяют автомобильный или водяные аспираторы различных конструкций.
Поглощение газов и паров в жидкие поглотительные среды. К жидким поглотительным средам относятся дистиллированная вода, органические растворители, специальные поглотительные растворы.
Поглощение газов и паров может происходить путем растворения их в жидкой среде или в результате взаимодействия токсического вещества с поглотительным раствором. Если поглощение основано на растворении вещества в растворителе, то такое поглощение может быть отнесено к малоэффективным. Обычно поглощение токсического вещества происходит при малых скоростях протягивания исследуемого воздуха и интенсивном охлаждении.
Увеличение скорости отбора проб, а также повышение температуры ведет к улетучиванию как растворителя, так и исследуемого вещества. В таком случае отбор среднесуточных проб возможен с перерывами 6 — 24 раза в отдельные поглотительные приборы с индивидуальным анализом (третий вариант). Если поглощение токсических веществ проводят в поглотительные растворы, содержащие соответствующие реактивы, то в процессе поглощения происходит реакция взаимодействия его с реактивом, обычно с образованием нелетучего соединения, чем обеспечивается более полное поглощение.
Следует учитывать, что во время отбора проб, особенно среднесуточных, происходит испарение растворителя, при этом объем поглотительной среды уменьшается. Во избежание изменения объема пробы периодически в процессе отбора пробы объем поглотительной жидкости доводят до первоначального уровня растворителем, используемым при анализе.
Эффективность поглощения вещества достигается возможно большим контактом его с поглощающей жидкой средой. С этой целью употребляются поглотительные приборы различных конструкций.
Поглотительные приборы с пористой стеклянной пластинкой (рис. 3) представляют собой V-образные трубки с впаянными в виде пластин стеклянными фильтрами N 1 и 2. Стеклянные фильтры сделаны из особо приготовленной спекшейся массы стекла с различными величинами пор.
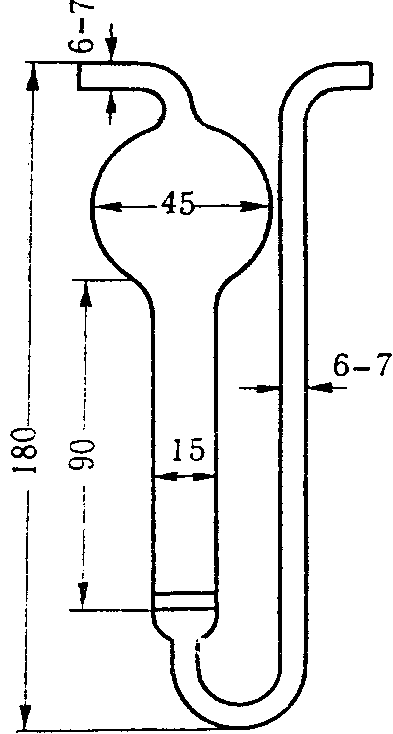
Рис. 3. Поглотительный прибор
с пористым стеклянным фильтром
N 1 — ПС-1 диаметр пор 90 — 150 мк, N 2 — ПС-2 диаметр пор 40 — 90 мк. Сопротивление фильтров устанавливают, наливая воду на фильтр высотой 5 мм, и протягивают воздух до появления первых пузырьков. Для ПС-1 сопротивление должно быть 25 — 15 мм рт. ст., для ПС-2 55 — 25 мм рт. ст. Диаметр фильтров 15 и 10 мм, толщина 2 — 3 мм (ГОСТ 9775-61). Используются поглотительные приборы емкостью 2 и 6 мл (малая и большая модель, диаметр пористой пластинки от 10 мм и более). В поглотительный прибор вносят жидкость, через которую протягивают исследуемый воздух. Воздух при помощи впаянной стеклянной пористой пластинки разбивается на множество мелких пузырьков, чем обеспечивается большая поверхность соприкосновения с поглотительной средой. Поглотительные приборы, снабженные пористой пластинкой, относятся к эффективным, обеспечивающим интенсивное поглощение, позволяющим проводить отбор проб со скоростью до 2 — 3 л/мин.
Поглотительные приборы Зайцева, Полежаева, Рыхтера (рис. 4) представляют собой стеклянные сосуды, внутри которых проходит цилиндрическая трубка, переходящая в некоторых случаях в расширение. В поглотительные сосуды вносят жидкую поглотительную среду по 2 — 10 мл. При применении поглотительных приборов Зайцева воздух протягивают со скоростью до 1 л/мин.; поглотительные приборы Полежаева обеспечивают поглощение при скорости протягивания воздуха до 0,5 л/мин., а приборы Рыхтера — до 10 л/мин.
Поглотительный прибор Гернет (рис. 5) представляет собой стеклянный грушевидной формы сосуд с распылителем типа форсунки. Воздух поступает в распылитель со скоростью 10 — 15 л/мин. При этом поглотительная среда распыляется внутри прибора и достигается большая поверхность соприкосновения жидкой среды с воздухом. Выводится воздух из прибора через верхнюю трубку, снабженную ловушкой со стеклянными бусами, служащими для улавливания брызг.
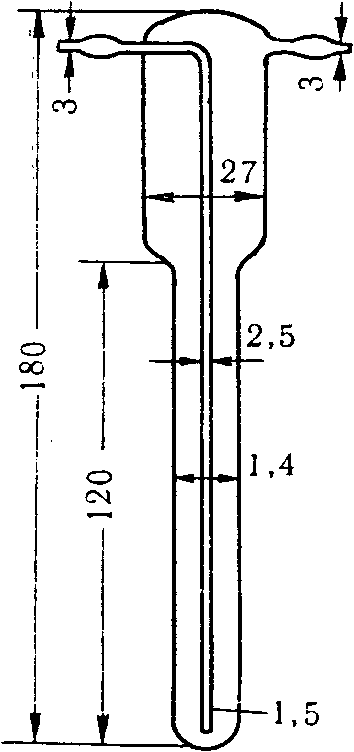
А
Б
В
Рис. 4. Поглотительные приборы Зайцева (А),
Рыхтера (Б), Полежаева (В)
Рис. 5. Поглотительный прибор Гернет
Поглощение газов и паров твердыми зерненными сорбентами [15, 16]. К твердым поглотительным средам относят зерненные сорбенты. При исследовании атмосферного воздуха чаще всего используют силикагель и активные угли. Твердые зерненные сорбенты характеризуются рядом положительных свойств — они удобны при хранении и транспортировке, обладают повышенными сорбционными свойствами при пониженных температурах.
Силикагель — пористое тело с сильно развитой поверхностью, зерна его характеризуются механической прочностью. Силикагель обладает ценным качеством — способностью сорбировать вещества и после специальной обработки восстанавливать свои первоначальные свойства. В зависимости от величины пор и формы зерен силикагель разделяют на мелкопористый, крупнопористый, кусковой и гранулированный.
В гигиенической практике обычно используют кусковой (как мелкопористый, так и крупнопористый) силикагель. Некоторые сведения, касающиеся кускового силикагеля, приведены в табл. 2 <1>.
———————————
<1> 1 Гранулированный силикагель содержит упрочняющую зерна добавку — 4 — 10% алюминия (ГОСТ 3956-54, силикагель).
Таблица 2
КУСКОВОЙ СИЛИКАГЕЛЬ
|
Марки силикагеля |
Величина зерен, мм |
|
КСМ — крупный силикагель мелкопористый |
2,7 — 7 |
|
ШСМ — шихта силикагель мелкопористый |
1,5 — 3,5 |
|
МСМ — мелкий силикагель мелкопористый |
0,25 — 3,5 |
|
АСМ — активированный силикагель мелкопористый |
0,2 — 0,5 |
|
КСК — крупный силикагель крупнопористый |
2,7 — 7 |
|
ШСК — шихта силикагель крупнопористый |
1,5 — 3,5 |
|
МСК — мелкий силикагель крупнопористый |
0,25 — 3,5 |
|
АСК — активированный силикагель крупнопористый |
0,2 — 0,5 |
Для поглощения токсических веществ чаще всего применяют силикагель с величиной частиц 0,25 — 2,00 мм, поэтому крупный силикагель, например КСМ, ШСМ, МСМ, КСК, ШСК, МСК, размельчают в ступке и, применяя соответствующий размер сита, готовят зерна необходимой величины.
Если силикагель окрашен (обычно за счет содержания железа), то его обрабатывают горячей соляной кислотой (удельный вес 1,19) и далее 10% раствором соляной кислоты до получения неокрашенного раствора. Затем силикагель промывают водой до удаления иона хлора (по реакции с азотнокислым серебром), высушивают при 100 °С и активируют при 200 — 150 °C в течение часа в сушильном шкафу. Хранят силикагель в закрытых склянках.
Силикагель перед употреблением проверяют на отсутствие вещества, подлежащего определению.
Температура в пределах +/- 40° и относительная влажность в интервалах 50 — 85% практически не влияют на сорбционную способность силикагеля.
В ряде случаев для поглощения токсических веществ применяют активные угли. Уголь, как и силикагель, представляет собой пористое тело, состоящее из углерода. Угли типа Г, АГ-5, АГ-3 активные, гранулированные, газовые, поглощающие пары и газы вследствие сильно развитой удельной поверхности. Уголь, как и силикагель, перед употреблением проверяют на отсутствие вещества, которое определяют, т.е. проводят контрольный опыт.
Для сорбции токсических веществ на твердых зерненных сорбентах последние помещают в специальные трубки или поглотительные приборы. Поглощение веществ проводят в неподвижном или «кипящем» слое.
При поглощении токсического вещества на неподвижный слой сорбент помещают в трубки различных конфигураций, иногда с перемежающимися сферическими расширениями (рис. 6). Сорбент в количестве до 5 см3 помещают в трубки, в концы которых закладывают стеклянную вату или спирали из проволоки для закрепления зерен. Ввиду большого сопротивления в таких трубках до 300 мм вод. ст. исследуемый воздух протягивают со скоростью лишь до 2 л/мин. и применяют мощные аспирационные устройства.
Рис. 6. Трубка со сферическими расширениями
для твердого сорбента
Поглощение токсических веществ из атмосферного воздуха можно проводить на кипящий слой зерненного сорбента.
При этом целесообразно использовать силикагель, так как его зерна обладают механической прочностью. Зерна силикагеля помещают в поглотительный прибор и через слой протягивают воздух. По мере увеличения скорости подачи воздуха, а следовательно, и его объема через слой сорбента этот слой значительно увеличивается, твердые частицы перестают касаться друг друга и приобретают подвижность. Образуется кипящий слой, обусловливающий эффективность перемешивания и контакт с исследуемым веществом. Отбор проб проводят со скоростью до 12 л/мин., при этом сопротивление кипящего слоя сравнительно невелико (60 мм вод. ст.).
Для отбора проб воздуха на кипящий слой используются поглотительные приборы Яворовской и Московского научно-исследовательского института гигиены имени Ф.Ф. Эрисмана и др. (рис. 7). Через боковую трубку поглотительных приборов при помощи стеклянной воронки вводят до 5 мл зернистого сорбента. С целью устранения переброса частиц силикагеля в поглотительных приборах предусмотрены вогнутые вглубь шипы.
Переброс зерен страхуется последовательным присоединением специального прибора, показанного на рис. 7. По окончании отбора проб переброшенные частицы силикагеля присоединяются к общей массе. Токсические вещества, поглощенные на сорбент, десорбируют путем экстрагирования соответствующими растворителями или растворами, в отдельных случаях десорбцию веществ осуществляют выдуванием с последующим поглощением в соответствующий раствор.
Поглощение аэрозолей. Для поглощения аэрозолей из воздуха используют фильтрующие материалы: плотные бумажные фильтры (беззольные), фильтры из тонких волокон и др., которые в виде дисков помещают в специальные патроны. Бумажные фильтры, обладая задерживающей способностью, оказывают большое сопротивление. Беззольный бумажный фильтр (синяя лента) с рабочей поверхностью 18 кв. см при скорости протягивания воздуха до 10 л/мин. имеет сопротивление до 300 мм вод. ст.
Рис. 7. Поглотительные приборы для отбора проб в кипящий
слой Московского научно-исследовательского института имени
Ф.Ф. Эрисмана (А), Яворовской (Б), для задерживания
переброшенных частиц (В)
В практике исследования воздушной среды для поглощения аэрозолей широко используются аналитические фильтры аэрозольные (АФА).
Фильтры АФА обладают высокой задерживающей способностью и практически полностью задерживают аэрозоли размером 0,1 — 0,2 мк при скорости протягивания до 100 л/мин. Фильтр обладает небольшим собственным весом, негигроскопичен (гидрофобен), стоек к химически агрессивным средам, растворим в ацетоне, дихлорэтане; пары и газообразные примеси фильтр АФА не задерживает. Фильтры АФА обладают сравнительно небольшим сопротивлением, например фильтр с рабочей поверхностью 18 кв. см имеет сопротивление 20 — 40 мм вод. ст. при скорости протягивания воздуха 20 л/мин.
Фильтры АФА хранят в закрытом виде при температуре не выше 40 °C в сухом помещении, в отсутствие паров масел, ацетона, дихлорэтана.
Осуществляется массовый выпуск фильтров АФА-В-10, АФА-В-18 для весового анализа с рабочей поверхностью 10 и 18 кв. см, АФА-XII-18 — перхлорвиниловые, предназначенные для химического анализа, и др. [8, 28].
Патроны представляют собой воронку, в широкой части которой при помощи специального кольца герметизируется фильтр. Ширина выреза в патроне соответствует рабочей поверхности фильтра. На рис. 8 указаны патроны и фильтры, используемые для отбора проб на аэрозоли; на рис. 9 показан патрон, используемый для задержания сажи. В отдельных случаях для поглощения аэрозолей применяют фильтры, предназначенные для отбора проб пыли, имеющие большую рабочую поверхность.
Рис. 8. Фильтр и патрон для отбора проб аэрозолей
А — общий вид и основные элементы фильтра;
Б — патрон для фильтров, имеющих рабочую
поверхность 18 и 20 кв. см
Рис. 9. Патрон для задерживания сажи
(рабочая поверхность 22 кв. мм)
Аспирационные приборы
Отбор проб в жидкие, твердые поглотительные среды, а также на фильтры проводят динамическим аспирационным способом при помощи специальных приборов. Такими приборами являются водяные аспираторы, электроаспираторы. Применяют и другие аспирационные устройства. Например, в практике широко используются автомобильные аспираторы, в отдельных случаях пользуются баллонами со сжатым воздухом, водоструйным насосом и т.д.
При этом объем исследуемого воздуха определяют при помощи самих водяных аспираторов или с применением измерительной аппаратуры.
Простейшими приборами для отбора проб воздуха являются водяные аспираторы, работающие по принципу сообщающихся сосудов.
Объем вытекаемой воды соответствует количеству воздуха, протянутого через поглотительный прибор (рис. 10).
Отбор круглосуточных проб проводится водяным аспиратором системы Рязанова (рис. 11). Этот аспиратор состоит из двух баков, расположенных один под другим. Емкость баков до 500 л. Баки сообщаются между собой через трубку с краном, которым регулируется скорость вытекания воды из верхнего бака в нижний.
Рис. 10. Водяной аспиратор, изготовленный из двух бутылей
1 — бутыль; 2 — пробка; 3 — длинная трубка — сифон;
4 — короткая трубка
Рис. 11. Водяной аспиратор системы Рязанова
Отбор проб проводят со скоростью до 500 л в сутки, около 10 — 20 л в час. Зимой аспиратор наполняют 26% раствором поваренной соли.
Отбор проб можно проводить, используя автомобильный двигатель как аспиратор, а в зимнее время как средство обогрева поглотительных приборов, заполненных жидкой поглотительной смесью.
Для отбора проб предложено специальное приспособление к санитарной автомашине УАЗ-450а, ПАЗ-653 [10].
Аспирация исследуемого воздуха осуществляется при помощи всасывающего коллектора двигателя и шланга. К шлангу присоединена съемная панель, на которой размещены системы, состоящие из реометров и поглотительных приборов (рис. 12). При работе двигателя аспирируется воздух со скоростью от 1 до 100 л/мин.
Рис. 12. Схема съемной приборной панели
1 — место крепления; 2 — полка для реометров; 3 — реометры;
4 — место крепления; 5 — гребенка; 6 — крепление
поглотительных приборов; 7 — 13 — место крепления
теплообменника; 8 — зажимы; 9 — шланги; 10 — всасывающий
коллектор двигателя; 11 — поглотительные приборы;
12 — воронка, через которую поступает исследуемый воздух
Для отбора проб воздуха широко применяют электроаспираторы, работающие от электролиний; вдали от электросети используются электроаспираторы, работающие на аккумуляторах.
Для отбора проб используют и бытовые пылесосы различной конструкции, при этом удобно пользоваться специальной приставкой [27].
На рис. 13 схематически изображена приставка и дан общий вид установки. Приставка представляет собой дюралюминиевую трубку с расширением в центральной части, где имеется 8 патрубков одинакового диаметра (1). Воздух протягивают со скоростью до 10 л/мин. С одной стороны трубки присоединен еще один патрубок большего диаметра (2), соединенный с винтом (3), которым регулируется степень разрежения в патрубках. Вращением винта прикрывают величину отверстия патрубка (4) и регулируют скорость отбора. К патрубкам через резиновые шланги присоединяют реометры и далее поглотительные приборы.
Рис. 13. Установка для отбора проб
А — приставка к пылесосу; Б — общий вид
Для отбора проб атмосферного воздуха довольно широко применяют универсальный электроаспиратор Мигунова-Кабанова (УАМК-3), а также стационарные электроаспираторы — аэрозольный ингалятор АИ-1, портативный аспиратор с аккумуляторными батареями Института общей и коммунальной гигиены имени А.Н. Сысина АМН СССР. Используются также автоматические и неавтоматические электроаспираторы, работающие на переменном токе и аккумуляторах, предложенные Л.Ф. Качором (ЛК-1, ЛК-2, ЛК-3).
ЛК-1 — переносной прибор, может быть установлен на любой автомашине, работает от аккумулятора напряжением 12 В, мощностью 55 Вт. Прибор снабжен ротаметрами, штуцерами для присоединения поглотительных приборов, рассчитан на непрерывное действие в течение 40 мин., после чего выключается для охлаждения. ЛК-2 и ЛК-3 — автоматические стационарные приборы, работающие на переменном токе. ЛК-2 предназначен для отбора разовых проб, снабжен несколькими ротаметрами, работает по заданной программе, позволяет отбирать пробы с интервалами. Прибор может работать и неавтоматически. ЛК-3 позволяет отбирать среднесуточные пробы, работает автоматически по заданной программе. Одновременно можно отбирать три пробы по трем каналам. В сутки по одному каналу проходят до 24 проб, по трем — до 72 [17, 18].
На рис. 14 представлен электрический аспиратор Качора.
Рис. 14. Электроаспиратор Качора
1 — штатив для поглотительных приборов; 2 — ротаметры;
3 — регуляторы скоростей; 4 — место освещения шкал
ротаметров; 5 — шланги; 6 — воздухосборник;
7 — электромотор; 8, 10 — места смазки;
9 — воздуходувка
Аспирационный отбор проб атмосферного воздуха проводят в различные сезоны года, при повышенных и пониженных температурах. Он связан с протягиванием значительных объемов воздуха. Если поглотительной средой служит жидкость, то при отборе проб при повышенных температурах замечено значительное испарение растворителя, а следовательно, и уменьшение объема поглотительного раствора. Испарение растворителей значительно устраняется применением охладительных смесей, в которые помещают поглотительные приборы.
При отборе проб в условиях пониженных температур часто поглотительные жидкости замерзают. Отбор проб при отрицательных температурах можно проводить, обогревая поглотительные приборы электролампой или простейшим обогревателем, позволяющим в отсутствие электроэнергии поддерживать необходимую температуру.
Обогреватель (рис. 15) представляет собой пластмассовую коробку (а), снабженную двойными стенками. Высота его 180 мм, длина 105 мм, ширина 85 мм. В пространстве между стенками помещен теплоизолирующий слой (б). В верхней части имеется отверстие (в), которое закрывается резиновой пластинкой (г). В резиновой пластинке высверлены отверстие (д) и разрез, куда вставляют поглотительный прибор (е) [12].
Рис. 15. Обогреватель поглотительных приборов
Рис. 16. Схема присоединения поглотительных приборов,
фильтров при измерении сопротивления
А — электроаспиратор; Б — реометр; В — манометр,
Г — место выступления воздуха
Перед отбором обогреватель заполняют водой, нагретой до 20 — 30 °C. При отборе проб со скоростью 1 — 2 л/мин. температура поглотительной жидкости поддерживается до 18 — 20° в течение 2 ч, если температура наружного воздуха находится в пределах 20 °C.
При отборе проб аспирационным методом обычно учитывают сопротивление поглотительных приборов, наполненных раствором или твердым сорбентом, также и фильтров, помещенных в патрон. Измерение сопротивления проводят с помощью водяного манометра, по схеме, указанной на рис. 16.
Отбор проб атмосферного воздуха
в сосуды ограниченной емкости
Отбор проб в ограниченные емкости обычно проводят в тех случаях, когда необходимо установить количество веществ, загрязняющих атмосферный воздух, за короткий промежуток времени. Воздух отбирают в сосуд, объем которого известен. Анализ проводят в малом объеме воздуха, в связи с чем могут быть применены лишь высокочувствительные методы.
Для отбора проб воздуха используют газовые пипетки емкостью до 1 л, а также бутыли различной емкости с пробками и трубками — одной длинной, доходящей до дна, другой короткой (рис. 17, А). Отбор проб может быть осуществлен различными способами: выливанием жидкости, обменным способом, способом вакуумирования воздуха. При использовании способа выливания жидкости емкость предварительно заполняют соответствующим раствором, не реагирующим с исследуемым веществом. В том месте, где берут пробу, жидкость выливают и емкость заполняют исследуемым воздухом. Регулируя скорость вытекания жидкости, удлиняют или укорачивают время отбора пробы.
Применяя обменный способ отбора, пипетку или бутыль присоединяют к аспиратору и на месте отбора пробы емкость продувают десятикратным объемом исследуемого воздуха. Во избежание конденсации паров исследуемого вещества на стенках сосуда продувание ведут со скоростью не менее 2 л/мин.
При отборе проб вакуумным методом применяют сосуды из толстостенного стекла, защищая их чехлом из металлической сетки (рис. 17, 1). Газовую пипетку или бутыль присоединяют к тройнику вакуум-манометра со стороны закрытого колена. Второй конец тройника присоединяют к насосу В (3). Откачивают воздух из емкости до остаточного давления 10 — 15 мм рт. ст. Остаточное давление можно измерить и при помощи открытого манометра, при этом во время откачивания воздуха из емкости один конец ее присоединяют к вакуум-насосу, а второй — к вакуум-манометру (рис. 17, 2). При откачивании воздуха из бутыли ее присоединяют к одной стороне манометра при помощи тройника, а второй конец тройника — к вакуум-насосу. Отбор проб осуществляется путем открывания сосуда в месте исследования атмосферного воздуха.
Рис. 17. Приспособления для отбора проб
в сосуды ограниченной емкости
А — бутыль и газовая пипетка; Б — заполнение пипетки
жидкостью; В — схема вакуумирования воздуха из емкости
ИЗМЕРЕНИЕ ОБЪЕМА ИССЛЕДУЕМОГО ВОЗДУХА
Для учета объема и скорости аспирируемого воздуха применяют реометры и другие приборы.
Реометр состоит из стеклянной V-образной манометрической трубки (рис. 18), части которой соединены между собой горизонтальной трубкой, в которую впаяна диафрагма. Манометрической жидкостью является очищенный керосин (удельный вес 0,82) или вода. При пропускании воздуха в той части манометрической трубки, которая находится до диафрагмы, создается давление, а в трубке, находящейся после диафрагмы, — разрежение, способствующее поднятию уровня манометрической жидкости: этот уровень и соответствует объему проходящего воздуха.
Рис. 18. Приборы, служащие для измерения
скорости прохождения исследуемого воздуха
А — реометр; Б — реометр с поворотными
диафрагмами (1а — место входа воздуха; 2 — шкала;
3 — диафрагма); В — пневмометр
Применяют и реометры с поворотными диафрагмами. Меняя диафрагмы, можно проводить измерение объема воздуха от 0,6 до 80 л/мин. Манометрической жидкостью служит вода. К реометру приложены калибровочные графики, по которым определяют скорость протянутого воздуха в единицу времени.
Могут быть использованы также и сухие ротаметры, представляющие собой вертикально расположенную стеклянную трубку в виде усеченного конуса. Внизу трубки на специальном стержне помещен поплавок строго определенного веса. Поток воздуха поднимает поплавок на уровень, соответствующий скорости движения воздуха (рис. 18). Объем воздуха, взятого для анализа, равен произведению скорости протягивания воздуха в минуту на время протягивания.
При отборе проб отмечают температуру воздуха и атмосферное давление в миллиметрах ртутного столба. В зависимости от сезона года эти данные неидентичны. Для получения сопоставимых результатов объем исследуемого воздуха приводят к нормальным условиям, выражая результат при 0° и давлении 760 мм рт. ст. по формуле:
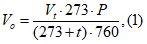
где Vt — объем взятого воздуха при температуре в точке отбора пробы;
Р — атмосферное давление в миллиметрах ртутного столба; t — температура воздуха в местах отбора проб.
Приведенной формулой можно пользоваться при расчете объема воздуха, отобранного аспирационным, обменным способами и способом выливания жидкости.
При вакуумном отборе проб регистрируется остаточное давление в емкости вакуум-манометром или открытым манометром. При использовании вакуум-манометра объем воздуха приводят к нормальным условиям по формуле:
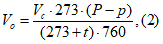
где Vс — объем сосуда; Р — атмосферное давление в миллиметрах ртутного столба; р — остаточное давление в сосуде, отмеченное вакуум-манометром, в миллиметрах ртутного столба, t — температура воздуха в месте отбора проб.
При использовании открытого манометра объем воздуха приводят к нормальным условиям по формуле:
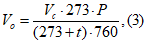
где Vс — объем сосуда; t — температура воздуха у места отбора проб; Р — показание открытого манометра.
Атмосферное давление измеряют в миллиметрах ртутного столба и миллибарах. Бар — давление, равное миллиону дин; 1 мб соответствует 1/1000 бар; 1000 мб соответствует 750, 38 мм рт. ст. Коэффициент пересчета с 1 мм рт. ст. на 1 мб равен 1,33.
Расчет объема воздуха, необходимого для анализа [5, 6]
Концентрации вредных веществ в атмосферном воздухе выражают в миллиграммах на 1 м3 воздуха. Минимальная концентрация токсического вещества, которая может быть определена, зависит от чувствительности применяемого метода. При одновременном присутствии токсических веществ, характеризующихся суммацией действия, когда сумма их концентраций не должна превышать единицы, необходимо определять концентрации веществ ниже предельно допустимых норм, что связано с протягиванием более значительных объемов исследуемого воздуха. Расчет требуемого объема воздуха для анализа проводят по формуле:
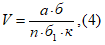
где а — чувствительность определения в микрограммах; б — общий объем пробы в миллилитрах; б1 — объем пробы, взятой для анализа, в миллилитрах; n — предельно допустимая концентрация в миллиграммах на 1 м3 воздуха; к — заданная доля предельно допустимой концентрации.
Расчет анализа проводят по формуле:
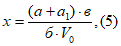
где х — концентрация искомого вещества в миллиграммах на 1 м3 воздуха; а — количество искомого вещества в одном поглотительном приборе, патроне, в микрограммах; а1 — количество искомого вещества во втором поглотительном приборе, патроне, в микрограммах; б — объем пробы, взятой для анализа, в миллилитрах; в — объем всей пробы в миллилитрах; V0 — объем протянутого воздуха, приведенного к нормальным условиям, в литрах.
ПРИГОТОВЛЕНИЕ СТАНДАРТНЫХ РАСТВОРОВ
Стандартным раствором называется раствор, содержащий определенное количество вещества в единице объема. Обычно готовят стандартный раствор из вещества, которое определяют в воздухе, или готовят из соединения, в состав которого входит ион искомого вещества. Первоначально готовят исходный стандартный раствор с содержанием вещества порядка десятых миллиграмма в 1 мл. Перед употреблением готовят рабочие стандартные растворы с содержанием сотых, тысячных долей миллиграмма в 1 мл.
Растворителем для приготовления стандартных растворов, как правило, служит поглотительная жидкость, применяемая для поглощения искомого вещества из воздуха.
Готовят стандартные растворы из жидких и твердых веществ в мерных колбах емкостью 100 мл и менее.
Для приготовления стандартного раствора из жидких веществ жидкость предварительно перегоняют и отбирают фракцию погона, отвечающую точке кипения вещества. Первые и последние погоны не используют для приготовления стандартных растворов. Для получения исходного стандартного раствора в мерную колбу вносят 2 — 5 мл растворителя, закрывают пробкой и взвешивают на аналитических весах.
Затем добавляют несколько капель исследуемого вещества и снова взвешивают. По разности весов определяют навеску вещества. Объем доводят до метки, перемешивают содержимое и рассчитывают содержание вещества в 1 мл раствора. Рабочие стандартные растворы готовят соответствующим разбавлением исходных. Вещество для приготовления стандартных растворов из твердых веществ предварительно очищают, перекристаллизовывают или отгоняют (например, фенол). Берут точную навеску и растворяют в мерной колбе.
Если готовят стандартный раствор из соли, то учитывают содержание кристаллизационной воды.
СТАТИСТИЧЕСКАЯ ОБРАБОТКА РЕЗУЛЬТАТОВ АНАЛИЗА
При количественном определении токсических веществ в воздухе с целью повышения точности, получения объективной оценки и улучшения качества анализа среди других приемов следует использовать и методы математической статистики. В условиях проведения анализа и при разработке методов определения малых количеств токсических веществ в воздухе приходится ограничиваться малым числом наблюдений. Поэтому для подтверждения статистической достоверности полученных данных используют формулы, пригодные для «малой выборки».
Обычно при анализе выполняют не менее трех — пяти параллельных определений и данные оцениваются как случайная выборка из генеральной совокупности. При обработке результатов наблюдений можно пользоваться следующей схемой. В качестве примера взято определение сернистого газа в воздухе. При анализе получены данные — 10; 12; 10; 12 и 11 мкг. Приведенные величины, будучи расположены в нарастающем порядке, т.е. 10; 10; 11; 12; 12, составляют вариационный ряд, в котором каждое число является вариантой (V) этого ряда.
Находим среднюю арифметическую (М) данного ряда:
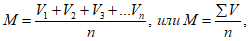
где V1, V2, V3 … до Vn есть значение вариант данного ряда; n — число наблюдений, т.е. число вариант; ![]() — знак суммирования.
— знак суммирования.
Таким образом, в нашем примере:
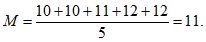
Следующим важным параметром вариационного ряда является среднее квадратичное отклонение (дельта), характеризующее степень различия между отдельными вариантами ряда:
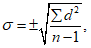
где d — разность между каждой вариантой ряда и полученной средней арифметической величиной, т.е. d = Vn — М.
В нашем примере:
V1 — М = 10 — 11 = -1
V2 — M = 10 — 11 = -1
V3 — M = 10 — 10 = 0
V4 — М = 12 — 11 = +1
V5 — M = 12 — 11 = +1
Как видно, отдельные величины отклоняются от среднего значения, причем они могут быть как положительными, так и отрицательными.
При правильном расчете сумма единичных отклонений должна быть равна 0, т.е. 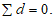 В нашем примере:
В нашем примере:
(-1 — 1 + 1 + 1) = 0.
Далее возводим каждое отклонение в квадрат и находим сумму квадратов отдельных отклонений:
|
d |
d2 |
|
-1 |
1 |
|
-1 |
1 |
|
0 |
0 |
|
+1 |
1 |
|
+1 |
1 |
|
|
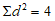
Таким образом:
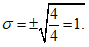
Хотя после извлечения корня квадратного получаются положительные и отрицательные величины, берут только положительное значение.
Для определения статистической достоверности средней арифметической величины используется средняя ее ошибка (mм):
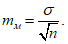
В нашем случае:
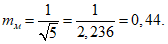
При числе наблюдений менее 30, т.е. когда n < 30, доверительные границы средней арифметической величины, по которым исследователь в каждом случае судит о достоверности полученной средней (М), пользуются выражением:
где величина множителя (t), называемая коэффициентом Стьюдента, определяется по таблице. Она показывает, во сколько раз надо увеличить среднюю ошибку, чтобы получить доверительные границы средней арифметической величины с желаемой степенью вероятности (95 или 99,9%).
В приведенном примере при вероятности в 95% t = 2,776.
Таким образом, доверительные границы полученной нами средней арифметической величины:
т.е. истинное значение М находится в пределах от 9,8 до 12,2.
Если исследователь считает установленные пределы колебаний средней величины допустимыми, то полученная величина М может считаться достоверной. В противном случае необходимо увеличить число и наблюдений.
Интересным показателем, с точки зрения аналитика, является величина коэффициента вариации ![]() Коэффициент вариации есть отношение среднего квадратичного отклонения
Коэффициент вариации есть отношение среднего квадратичного отклонения ![]() к средней величине (М), выраженное в процентах:
к средней величине (М), выраженное в процентах:
В данном примере коэффициент вариации равен 9,8%. 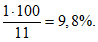
ФОРМА ЗАПИСИ РЕЗУЛЬТАТОВ
|
Наименование статистической характеристики |
Результаты серии опытов |
|
Число определений (n) |
5 |
|
Средняя величина (М) |
11 |
|
Стандартное отклонение отдельных результатов |
1,2 |
|
Средняя ошибка средней величины (tmм) |
0,44 |
|
Коэффициент вариации |
9,8 |
ЛИТЕРАТУРА
1. Алексеева М.В. Методы анализа. В кн.: Определение атмосферных загрязнений. М., «Медгиз», 1963.
2. Алексеев Р.И., Коровин Ю.И. Вычисления, производимые при выполнении анализа. В кн.: Руководство по вычислению и обработке результатов количественного анализа. М., «Атомиздат», 1972.
3. Агаджанян И.А. Человек, атмосфера и солнце. М., «Знание», 1968.
4. Атласов А.Г. Определение оптимального объема воздуха при отборе проб воздуха на вредные вещества. Гиг. и сан., 1965, 7, 2.
5. Атласов А.Г. Справочник по отбору проб воздуха на токсические примеси. Проектный институт «Проектпромвентиляция». Ч. 1. М., 1964.
6. Быховская М.С., Гинзбург С.Л., Хализова О.Д. Отбор проб воздуха. В кн.: Определение вредных веществ в воздухе. М., «Медицина», 1966.
7. Бабенко А.С., Володченко Т.Т. Применение цилиндрических кювет для снижения определяемого минимума при измерениях на ФЭК-М. Известия высших учебных заведений. Химия и химическая технология, 1968, в. 10, т. II.
8. Басманов П.И., Борисов Н.Б. В Бр. Фильтры АФА. Каталог-справочник. М., «Атомиздат», 1970.
9. Бланк А.Б. Простое приспособление для снижения определения при измерении на фотоэлектроколориметре ФЭК-М, ФЭК-Н. Журнал аналитической химии АН СССР, 1962, т. 17, в. 8.
10. Близнев В.И. Приспособление к санитарной машине УАЗ-450а для отбора атмосферного воздуха. Гиг. и сан., 1964, 5.
11. «В интересах народа». Передовая статья газеты «Правда» от 23 сентября 1972 г.
12. Гильденскиольд Р.С., Рихтер Б.В. Полевой обогреватель для поглотителей при отборе проб воздуха. Гиг. и сан., 1968, 2.
13. Доерфель К. Приемы вычислений. В кн.: Статистика в аналитической химии. М., «Мир», 1969.
14. Инструктивно-методические указания по организации исследования атмосферного воздуха. Медицинская литература. М., 1963.
15. Качмар Е.Г., Хрусталева В.А. Применение твердых зерненных сорбентов при исследовании атмосферного воздуха. Гиг. и сан., 1969, 9.
16. Качмар Е.Г., Хрусталева В.А. Определение вредных веществ в атмосферном воздухе с применением твердых зерненных сорбентов. Московский научно-исследовательский институт гигиены им. Ф.Ф. Эрисмана. М., 1964.
17. Качор Л.Ф. Конструкция аспираторов для отбора проб пыли и газов в атмосферном воздухе. Гиг. и сан., 1950, 4.
18. Качор Л.Ф. Новая аппаратура для отбора проб атмосферного воздуха. Гиг. и сан., 1964, 9.
19. Коренман И.М. Введение. В кн.: Введение в количественный ультрамикроанализ. М., «Госхимиздат», 1968.
20. Коренман И.М. Некоторые особенности очень малых разбавленных растворов. В кн.: Аналитическая химия малых концентраций. М., «Химия», 1968.
21. Ноткин Е.Л. Математическая статистика в гигиенических исследованиях. Вопросы санитарной статистики. Ученые записки N 10 Московского научно-исследовательского института гигиены им. Ф.Ф. Эрисмана. М., 1962.
22. О мерах по дальнейшему улучшению охраны природы и рациональному использованию природных ресурсов. Постановление Верховного Совета СССР. Газета «Правда» N 265 от 21 сентября 1972 г.
23. О мерах по дальнейшему улучшению здравоохранения и развитию медицинской науки в стране. Постановление ЦК КПСС и Совета Министров СССР от 5 июля 1968 г. N 517.
24. Основы законодательства Союза ССР и союзных республик о здравоохранении. «Медицинская газета» от 23 декабря 1969 г.
25. Предельно допустимые концентрации вредных веществ в атмосферном воздухе населенных мест. Санитарные нормы проектирования промышленных предприятий. СН-245-71. М., 1972.
26. Решения XXIV съезда КПСС.
27. Семенов В.А., Попов Е.И. Приставка к пылесосу для отбора проб атмосферного воздуха. Гиг. и сан., 1965, 8.
28. Шпинькова Л.И. Фильтры АФА. М., «Изотоп», 1972.
Глава II
ГАЛОГЕНЫ И ГАЛОИДОВОДОРОДЫ
ФТОРИСТЫЙ ВОДОРОД
Бесцветный газ с резким запахом, хорошо растворимый в воде. Плотность по отношению к воздуху 0,713, температура кипения 19,5°, температура плавления -81°, температура замерзания -179°. Фтористоводородная (плавиковая) кислота, бесцветная жидкость, дымящая во влажном воздухе, смешивающаяся с водой в любых соотношениях, плотность 1,15, содержит 35,3% фтористого водорода; как и фтористый водород, реагирует со многими металлами и кремниевым ангидридом (SiO2), сохраняется в парафинированных сосудах.
Фтористый водород сильно раздражает верхние дыхательные пути, обладает также общей токсичностью.
Предельно допустимые концентрации газообразных фтористых соединений (HF, SiF4) (в пересчете на фтор): максимальная разовая 0,02 мг/ м3, среднесуточная 0,005 мг/ м3.
ОПРЕДЕЛЕНИЕ ФТОРИСТОГО ВОДОРОДА
С ТИТАНХРОМОТРОПОВЫМ РЕАКТИВОМ [6]
Принцип определения
При взаимодействии иона фтора с титанхромотроповым комплексом образуется титанфторидное соединение. Окраска раствора в зависимости от концентрации иона фтора изменяется от темно- до светло-розовой, малодиссоциируемого титанфторидного соединения.
Чувствительность определения 2,0 мкг иона фтора в анализируемом объеме раствора. Определению не мешают сернистый газ, серная кислота.
Реактивы
1. Поглотительный раствор — дистиллированная вода.
2. Стандартные растворы фторида натрия. Исходный стандартный раствор с содержанием в 1 мл 100 мкг иона фтора готовят растворением 0,0221 г перекристаллизованного фторида натрия в 100 мл воды в мерной колбе. Рабочий стандартный раствор с содержанием 10 мкг/мл иона фтора готовят разведением исходного стандартного раствора в 10 раз. Очистка фторида натрия — см. примечание.
3. Двуокись титана.
4. Сульфат титана.
Для получения сульфата титана 20 г пиросернокислого калия и 1 г двуокиси титана сплавляют в кварцевом или фарфоровом тигле емкостью 100 — 200 мл при температуре не выше 800° в течение нескольких часов до получения прозрачного сплава. После остывания сплав заливают 5% раствором серной кислоты и оставляют до следующего дня. Сернокислый титан растворяют при комнатной температуре в 500 мл 5% раствора серной кислоты, все время помешивая, так как растворение идет медленно. После растворения раствор фильтруют через бумажный фильтр, раствор должен быть прозрачным.
Проверяют содержание сульфата титана в 1 мл раствора весовым методом. Для этого в 3 стакана емкостью по 50 мл каждый приливают по 10 мл приготовленного раствора, добавляют по 2 мл 10% раствора хлористого аммония, 1 каплю метилового оранжевого и по каплям 25% раствор аммиака до слабого запаха. Титан выпадает в виде хлопьев гидроокиси титана.
Осадок отфильтровывают через беззольный фильтр и промывают 2% раствором азотнокислого аммония до удаления иона сульфата. Фильтры с осадками озоляют, прокаливают при температуре 1000 — 1100 °C и взвешивают двуокись титана. Определяют содержание двуокиси титана в растворе. Содержание должно быть около 2 мг двуокиси титана или около 6 мг сернокислого титана в 1 мл приготовленного раствора. Раствор годен к употреблению в течение нескольких лет. Его хранят в темном месте.
5. Пиросульфат калия (натрия).
6. Серная кислота, 5% раствор.
7. Аммиак, 25% раствор.
8. Нитрат аммония, 2% раствор.
9. Хромотроповая кислота или ее динатриевая соль. 20 мг кислоты растворяют в 10 мл воды перед использованием.
10. Монохлоруксусная кислота.
11. Едкий натр, 0,1 и 0,005 н. растворы.
12. Буферный раствор с pH 2,85. 0,95 г монохлоруксусной кислоты растворяют в 100 мл воды, к раствору приливают 50 мл 0,1 н. раствора едкого натра.
13. Составной раствор. В мерную колбу емкостью 100 мл вносят 4,8 мг (или 0,8 мл раствора) сернокислого титана, 10 мл раствора хромотроповой кислоты, 20 мл буферной смеси, доводят водой до метки и перемешивают. Составной раствор, окрашенный в вишневый цвет, годен к употреблению в течение месяца.
14. Парафин.
15. Метиловый оранжевый, 0,5% раствор.
Отбор пробы
Для определения разовой концентрации воздух со скоростью 8 — 10 л/мин. протягивают через 2 последовательно соединенные поглотительные прибора Рыхтера, содержащие по 6 мл воды. Внутреннюю поверхность первого поглотительного прибора парафинируют. Продолжительность отбора 20 — 30 мин.
Ход анализа
5 мл пробы из каждого поглотительного прибора переносят в пробирки и проверяют pH среды. Если раствор имеет кислую реакцию, то его нейтрализуют 0,005 н. раствором едкого натра с индикатором метиловым оранжевым. В фарфоровой чашке к 0,005 мл исследуемого раствора добавляют 1 каплю метилового оранжевого и, если раствор окрашивается в розовый цвет, приливают из микробюретки 0,005 н. раствор едкого натра до окраски, равной окраске контроля (вода с одной каплей индикатора). Зная, сколько потребовалось щелочи на 0,5 мл раствора, прибавляют к 5 мл 10-кратное количество щелочи, взбалтывают и берут 5 мл для анализа. Затем к пробам приливают по 3 мл составного раствора, хорошо перемешивают и через 10 мин. фотометрируют в кюветах с толщиной слоя 2 см при длине волны 490 нм по сравнению с контролем, который готовят одновременно и аналогично пробам. Содержание иона фтора в анализируемом объеме определяют по предварительно построенному калибровочному графику.
Для построения калибровочного графика готовят шкалу стандартов (табл. 3).
Таблица 3
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
|
Рабочий стандартный раствор, мл |
0 |
0,2 |
0,4 |
0,7 |
1,0 |
1,5 |
2,0 |
|
Вода, мл |
5 |
4,8 |
4,6 |
4,3 |
4,0 |
3,5 |
3,0 |
|
Содержание иона фтора, мкг |
0 |
2 |
4 |
7 |
10 |
15 |
20 |
С увеличением содержания иона фтора окраска растворов ослабевает. Все пробирки шкалы обрабатывают аналогично пробам и измеряют оптическую плотность.
Шкалой стандартов можно пользоваться для визуального определения, ее готовят в колориметрических пробирках одновременно с пробами. В пробирках с притертыми пробками шкала устойчива в течение месяца.
Расчет см. выше.
Примечания. 1. Очистка фторида натрия. В 1 л горячей воды растворяют 45 г фторида натрия, добавляют 15 г хлорида калия и оставляют на ночь. Раствор фильтруют и выпаривают в платиновой чашке на водяной бане примерно до 40 мл жидкости. Кристаллы фторида натрия отсасывают и промывают холодной дистиллированной водой до отрицательной реакции на ион хлора. Фторид натрия сушат при 120 — 150 °C и хранят в стеклянной банке с притертой пробкой.
2. Предложенный метод определения фтористого водорода может быть использован и для определения других неорганических газообразных соединений фтора.
ОПРЕДЕЛЕНИЕ ФТОРИСТОГО ВОДОРОДА С ЭРИОХРОМЦИАНИНОМ [5]
Принцип определения
Ион фтора в щелочной среде вызывает ослабление окраски соединения, образующегося при взаимодействии соединений фтора с эриохромцианином и хлористым цирконием.
Чувствительность определения 1 мкг в анализируемом объеме раствора.
Реактивы
1. Поглотительный раствор. 20 мл 0,1 н. раствора едкого натра разбавляют до 1 л дистиллированной водой.
2. Исходный стандартный раствор фтористого натрия.
0,1105 г фтористого натрия растворяют в дистиллированной воде и объем доводят водой до 500 мл. Рабочий стандартный раствор с содержанием 10 мкг иона фтора в 1 мл готовят разбавлением водой исходного раствора.
3. Едкий натр, 0,1 н. раствор.
4. Эриохромцианин-R.
0,108 г эриохромцианина-R растворяют в 100 мл дистиллированной воды.
5. Хлорокись циркония.
В небольшом количестве дистиллированной воды растворяют 125 мг хлорокиси циркония, затем добавляют 15 мл соляной концентрированной кислоты и объем доводят водой до 100 мл.
Отбор пробы
Для определения разовой концентрации исследуемый воздух со скоростью 3 — 5 л/мин. просасывают через поглотительный прибор Рыхтера, наполненный 10 мл поглотительного раствора.
Ход анализа
Содержимое поглотительного прибора доводят дистиллированной водой до первоначального объема. Переносят пробу в пробирку, добавляют 0,5 мл раствора эриохромцианина, 1 мл раствора хлорокиси циркония, взбалтывают и нагревают в течение 10 мин. при 20 °C. Затем окраски растворов фотометрируют при длине волны 530 нм в кювете с толщиной слоя 1 — 2 см по отношению к контролю, который готовят одновременно и аналогично пробе. Содержание иона фтора в анализируемом объеме раствора определяют по предварительно построенному градуировочному графику. Для построения градуировочного графика готовят шкалу стандартов (табл. 4).
Таблица 4
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
|
Рабочий стандартный раствор, мл |
1 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
2,0 |
|
Поглотительный раствор, мл |
10 |
9,9 |
9,8 |
9,6 |
9,4 |
9,2 |
9,0 |
8,0 |
|
Содержание иона фтора, мкг |
0 |
1 |
2 |
4 |
6 |
8 |
10 |
20 |
Шкалу стандартов обрабатывают аналогично пробе, измеряют оптическую плотность и строят график.
Расчет см. выше.
ХЛОР
Газ желтовато-зеленого цвета, обладает резким раздражающим запахом, плотность при 0° — 2,49 (по воздуху).
Хлор легко сжижается в желтовато-зеленую жидкость под давлением 6 атм., плотность жидкости 1,55 при 34,1°.
Растворим в воде, хлороформе, четыреххлористом углероде и других органических растворителях. Сорбируется активированным углем.
Хлор действует раздражающе на дыхательные органы и вызывает отек легких.
Предельно допустимые концентрации: максимальная разовая 0,10 мг/м3, среднесуточная 0,03 мг/м3.
ОПРЕДЕЛЕНИЕ ХЛОРА С МЕТИЛОВЫМ ОРАНЖЕВЫМ [2, 3]
Принцип определения
При взаимодействии хлора в кислой среде с метиловым оранжевым ослабляется розовая окраска раствора. Уменьшение интенсивности окраски пропорционально количеству хлора.
Чувствительность определения — 0,2 мкг в анализируемом объеме раствора.
Сернистый газ, окислы азота до 10 мг/м3, а озон в концентрациях до 5 мг/м3 не мешают определению.
Реактивы
1. Поглотительный раствор: готовят исходный раствор. В мерной колбе емкостью 1 л растворяют 0,1 г метилового оранжевого в 100 мл горячей воды, по охлаждении прибавляют 0,5 г бромида калия х.ч., 20 мл этанола, содержание колбы перемешивают и добавляют воды до метки.
Раствор устойчив.
Для приготовления рабочего поглотительного раствора исходный раствор разбавляют. В мерную колбу емкостью 100 мл вливают 4 мл 0,05 н. серной кислоты и 5 мл исходного раствора и доводят до метки водой; 25 мл этого раствора помещают в коническую колбу и титруют рабочим стандартным раствором хлора до лимонно-желтой окраски. На 25 мл рабочего поглотительного раствора должно расходоваться 2,5 — 3 мл рабочего стандартного раствора хлора.
Если на титрование расходуется больше или меньше стандартного раствора хлора, то необходимо поглотительный раствор соответствующим образом разбавить или приготовить более концентрированный раствор.
Хранить его следует в темном месте.
2. Стандартный раствор хлора.
Готовят исходный раствор: 5 г хлорной извести растворяют в 250 мл воды. Раствор оставляют в покое на несколько часов и прозрачный раствор фильтруют в мерную колбу емкостью 1 л, доводят до метки дважды перегнанной водой, переливают в темную склянку и определяют титр раствора титрованием его 0,02 н. раствором тиосульфата.
Перед применением раствора каждый раз определяют содержание хлора йодометрически. Для титрования берут 10 мл исходного раствора хлора в коническую колбу с притертой пробкой емкостью 250 мл, вливают 5 мл 10% серной кислоты, 60 мл воды, всыпают 1 г йодида калия, раствор взбалтывают и оставляют в темном месте на 5 — 10 мин. Затем титруют свежеприготовленным 0,02 н. раствором тиосульфата до образования бледно-желтой окраски, вливают в раствор 1 мл 0,5% крахмала и титруют до обесцвечивания. Содержание хлора определяют из расчета: 1 мл 0,02 н. раствора тиосульфата соответствует 0,709 мг хлора.
Рабочий стандартный раствор с содержанием 10 мкг/мл получают разбавлением исходного раствора. В мерную колбу емкостью 100 мл вливают такое количество исходного раствора, в котором содержалось бы 1000 мкг хлора, и доводят до метки дважды перегнанной водой. Перед применением проверяют содержание хлора йодометрически, титруют раствор 0,002 н. раствором тиосульфата, который готовят перед титрованием. 1 мл 0,002 н. раствора тиосульфата соответствует 0,0709 мг хлора.
3. Метилоранжевый, х.ч.
4. Калий бромид, х.ч.
5. Этанол 96°, х.ч.
6. Серная кислота, 0,05 н. раствор.
7. Хлорамин Б.
8. Йод возогнанный, растертый и высушенный.
9. Тиосульфат, 0,1 н. раствор.
Отбор пробы
а) Для определения разовой концентрации воздух со скоростью 0,5 л/мин. протягивают через поглотительный прибор с пористым фильтром N 1, содержащим 2,5 мл поглотительного раствора. Продолжительность отбора 20 — 30 мин. При изменении окраски поглотительного раствора отбор прекращают.
б) Для определения среднесуточной концентрации воздух со скоростью 5 л/ч протягивают в течение суток через поглотительный прибор с пористым фильтром N 1, содержащий 5 мл поглотительного раствора. При наполнении прибора отмечают уровень жидкости, который периодически добавляют водой до первоначального.
Допустимо проводить отбор с перерывами в 2 — 4 ч в один и тот же поглотительный прибор.
Ход анализа
Из поглотительного прибора жидкость выливают в колориметрическую пробирку. Одновременно строят шкалу стандартов (табл. 5). Пробу колориметрируют на белом фоне.
Таблица 5
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
|
Рабочий стандартный раствор, мл |
0 |
0,02 |
0,05 |
0,10 |
0,15 |
0,20 |
0,25 |
|
Поглотительный раствор, мл |
2 |
1,98 |
1,95 |
1,90 |
1,85 |
1,80 |
1,75 |
|
Содержание хлора, мкг |
0 |
0,20 |
0,50 |
1,0 |
1,5 |
2,0 |
2,5 |
Шкала сохраняется в течение месяца в темном месте.
Расчет см. выше.
Примечание. Можно концентрацию хлора установить фотометрически. Для этого готовят шкалу стандартов с содержанием 1, 2, 3 и 4 мкг хлора в объеме 5 мл. Измеряют величины оптических плотностей полученных растворов и строят калибровочный график. В качестве контроля используют поглотительный раствор, но предварительно измеряют его оптическую плотность по отношению к дистиллированной воде. Перед отбором проб проверяют оптическую плотность поглотительного раствора. Если величина оптической плотности изменилась, то раствор разбавляют водой или добавляют в него исходного поглотительного раствора до получения первоначальных данных.
ХЛОРИСТЫЙ ВОДОРОД И СОЛЯНАЯ КИСЛОТА
Хлористый водород — бесцветный газ с резким запахом, плотность по отношению к воздуху 1,26, вес 1 л его при 0 °C 1,6391.
Хлористый водород поглощается водой, один объем воды при комнатной температуре и атмосферном давлении может растворить 450 объемов хлористого водорода. Он растворяется в эфире и спирте. Во влажном воздухе хлористый водород образует аэрозоль соляной кислоты. Хлористый водород и аэрозоль соляной кислоты раздражают верхние дыхательные пути, вызывают воспаление слизистой оболочки глаз и помутнение роговицы.
Предельно допустимые концентрации: максимально разовая по молекуле — 0,2 мг/м3, по иону водорода — 0,006 мг/м3, среднесуточная по молекуле — 0,2 мг/м3, по иону водорода — 0,006 мг/м3.
ОПРЕДЕЛЕНИЕ ХЛОРИСТОГО ВОДОРОДА И СОЛЯНОЙ КИСЛОТЫ
С НИТРАТОМ СЕРЕБРА [1]
Принцип определения
При взаимодействии иона хлора с нитратом серебра образуется взвешенная муть. По интенсивности помутнения определяют содержание иона хлора.
Чувствительность определения — 2 мкг в анализируемом объеме. Соли других галоидов и синильной кислоты мешают определению.
Реактивы
1. Поглотительный раствор — дважды перегнанная вода.
2. Стандартный раствор хлористого водорода. Исходный раствор с содержанием 100 мкг/мл готовят растворением 0,0204 г хлорида калия в воде в мерной колбе емкостью 100 мл.
Рабочий стандартный раствор с содержанием 10 мкг/мл получают разбавлением исходного раствора в 10 раз.
3. Нитрат серебра, 1% раствор (хранят в темной склянке).
4. Азотная кислота, 10% раствор.
Все реактивы готовят на дважды перегнанной воде, свободной от иона хлора.
Отбор пробы
а) Для определения разовой концентрации протягивают воздух со скоростью 1 л/мин. через два поглотительных прибора с пористым фильтром N 1, содержащие по 6 мл дважды перегнанной воды. Продолжительность отбора 20 — 30 мин.
б) Для определения среднесуточной концентрации непрерывно в течение суток воздух со скоростью 10 л/ч протягивают через вышеуказанный поглотительный прибор, содержащий 6 мл поглотительного раствора. При наполнении прибора отмечают уровень жидкости, периодически добавляют водой до первоначального уровня.
Допустимо проводить отбор 6 — 12 раз с перерывом в 2 — 4 ч в один и тот же поглотительный прибор.
Ход анализа
В пробирку переносят 2,5 — 3 мл пробы, вливают 1 мл 10% азотной кислоты и 0,5 мл нитрата серебра. Содержимое пробы встряхивают, через 10 мин. помещают в кювету шириной 1 см и сравнивают с контролем с помощью фотоэлектроколориметра при длине волны 364 нм.
Содержание вещества в анализируемом объеме определяют по предварительно построенному калибровочному графику.
Для построения калибровочного графика готовят шкалу стандартов (табл. 6).
Таблица 6
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
|
Рабочий стандартный раствор, мл |
0 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
2,0 |
|
Поглотительный раствор, мл |
2,5 |
2,3 |
2,1 |
1,9 |
1,7 |
1,5 |
0,5 |
|
Содержание иона хлора, мкг |
0 |
2,0 |
4,0 |
6,0 |
8,0 |
10,0 |
20,0 |
Все пробирки шкалы обрабатывают аналогично пробе.
Шкалой стандартов можно пользоваться для визуального определения, ее готовят в колориметрических пробирках одновременно с пробами.
Расчет см. выше.
ОПРЕДЕЛЕНИЕ СОЛЯНОЙ КИСЛОТЫ ПО ИОНУ ВОДОРОДА [2, 4]
Принцип определения
Метод основан на измерении оптической плотности при длине волны 530 нм окрашенного водного раствора нацело диссоциированной кислоты с метиловым красным в интервале значения pH 4,7 — 5,3.
Чувствительность определения — 0,05 мкг ионов водорода в анализируемом объеме (5 мл).
Определению мешают основания и кислоты, реагирующие с метиловым красным в указанном интервале pH.
Присутствие CO2 в количестве 0,05 об.% и SO2 в количестве 0,5 мг/м3 не мешает определению.
Аппаратура
1. Спектрофотометр, позволяющий проводить измерение оптической плотности в видимой области спектра, или фотоэлектроколориметр.
2. Поглотительные приборы Зайцева.
Реактивы
1. Дистиллированная вода, ГОСТ 6709-53.
Ее проверяют добавлением к 5 мл воды 1 капли метилового красного и 1 капли 0,005 н. раствора соляной кислоты. При pH 5,8 — 6,6 раствор окрашивается в розовый цвет.
2. 0,1 н. раствор соляной кислоты. Перед употреблением готовят исходный 0,005 н. раствор соляной кислоты с содержанием 5 мкг/мл ионов водорода.
Рабочий стандартный раствор с содержанием 0,5 мкг ионов водорода в 1 мл готовят разбавлением исходного в 10 раз.
3. 0,01% раствор метилового красного.
Отбор пробы
Для определения разовой концентрации исследуемый воздух протягивают через поглотительный прибор, наполненный 6 — 10 мл дистиллированной воды, со скоростью 0,5 л/мин. в течение 20 мин.
По окончании отбора объем пробы доводят до первоначального.
Ход анализа
К 5 мл испытуемого раствора добавляют 0,2 мл раствора метилового красного, помещают в кювету шириной 1 — 2 см и через 1 — 1 1/2 мин. замеряют оптическую плотность по сравнению с контролем при длине волны 530 нм.
Содержание иона водорода в анализируемом объеме определяют по заранее построенному калибровочному графику. Для построения графика в пробирках готовят шкалу стандартов (табл. 7).
Таблица 7
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
|
Рабочий стандартный раствор, мл |
0 |
0,1 |
0,15 |
0,2 |
0,3 |
0,4 |
|
Раствор метилового красного, мл |
По 0,4 мл |
|||||
|
Дистиллированная вода, мл |
10 |
9,5 |
9,45 |
9,4 |
9,3 |
9,2 |
|
Содержание ионов водорода, мкг |
0 |
0,05 |
0,07 |
0,1 |
0,15 |
0,2 |
Содержимое пробирок перемешивают, измеряют оптическую плотность и строят график.
Расчет см. выше.
ЛИТЕРАТУРА
1. Алексеева М.В. Определение хлористого водорода и соляной кислоты. В кн.: Определение атмосферных загрязнений. М., «Медгиз», 1963.
2. Вильнер Т.А. К определению хлора с метиловым оранжевым в атмосферном воздухе. Гиг. и сан., 1972, 6.
3. Криворучко Ф.Д. Определение хлора в атмосферном воздухе. Гиг. и сан., 1953, 3.
4. Манита М.Д. Спектрофотометрический метод определения азотной и соляной кислот в присутствии нитратов и хлоридов в атмосферном воздухе. Гиг. и сан., 1964, 3.
5. Определение фтористого водорода с эриохромцианином. В сб.: СЭВ. Унифицированные методы определения атмосферных загрязнений. Ч. 1. М., 1970.
6. Панин К.П. Определение фтористого водорода (Фотометрический метод). Гиг. и сан., 1967, 12.
Глава III
СОЕДИНЕНИЯ СЕРЫ
СЕРНИСТЫЙ АНГИДРИД (СЕРНИСТЫЙ ГАЗ)
Бесцветный газ, с резким характерным запахом, который не поддерживает горение и сам не горит.
Плотность при нормальных условиях по отношению к воздуху 2,2630.
Вес 1 л газообразного SO2 2,9256 г.
Сернистый газ хорошо растворим в воде: в 100 г воды при 20 °C растворяется 10,5 г, при 10 °C — 15,4 г, при 0 °C в одном объеме воды — 70 объемов сернистого газа. Водный раствор имеет кислую реакцию.
Сернистый газ обладает восстановительными свойствами.
Действуя на организм, сернистый газ раздражает дыхательные пути, слизистые оболочки глаз, вызывает кашель.
Предельно допустимые концентрации: максимальная разовая доза 0,5 мг/м3; среднесуточная 0,15 мг/м3.
ОПРЕДЕЛЕНИЕ СЕРНИСТОГО АНГИДРИДА
С ФУКСИНФОРМАЛЬДЕГИДНЫМ РЕАКТИВОМ [1, 2]
Принцип определения
Сернистый ангидрид улавливают в раствор тетрахлормеркурата натрия с прибавлением комплексона (для предотвращения каталитического окисления). Образующееся соединение дает в кислой среде с фуксином и формальдегидом красно-фиолетовую окраску, интенсивность которой пропорциональна количеству сернистого газа.
Чувствительность определения — 0,3 мкг в анализируемом объеме раствора.
Определению не мешают хлор, озон в концентрациях, превышающих содержание сернистого ангидрида. Сероводород устраняют в виде сульфида ртути, отделяя его фильтрованием или центрифугированием.
Двуокись азота мешает определению.
Реактивы
1. Поглотительный раствор; 0,1 М раствор тетрахлормеркуроата натрия и 0,01% раствор комплексона III (динатриевой соли этилендиаминотетрауксусной кислоты) готовят путем растворения 27,2 г HgCl2, 11,7 г NaCl и 0,1 г комплексона III в 1 л дистиллированной воды.
2. Исходный стандартный раствор сульфита. 0,1968 г безводного сульфита натрия х.ч. или 0,1735 г пиросульфита калия (K2S2O5) растворяют в 1 л поглотительного раствора. В 1 мл раствора содержится 100 мкг сернистого газа.
3. Рабочий стандартный раствор. Разбавляют 5 мл исходного раствора до объема 100 мл поглотительным раствором. 1 мл такого раствора содержит 5 мкг сернистого газа.
4. Раствор фуксина: 0,65 г основного фуксина (кристаллического) х.ч. растворяют после размельчения в 22 мл этанола, разбавляют 100 мл дистиллированной воды, медленно и после охлаждения добавляют 60 мл концентрированной серной кислоты х.ч. и после охлаждения осторожно доводят водой до метки 1 л.
5. 2% раствор формальдегида х.ч. Для этого 5 мл 40% раствора формальдегида доводят водой до 100 мл.
6. Фуксинформальдегидный реактив: 10 частей свежепрофильтрованного раствора фуксина смешивают с 1 частью раствора формальдегида. Реактив применяют свежеприготовленный.
Отбор пробы
а) Для определения разовой концентрации протягивают воздух со скоростью 2 л/мин. через поглотительный прибор с пористым фильтром N 1, содержащий 6 мл поглотительного раствора. Продолжительность отбора 20 — 30 мин.
б) Для определения среднесуточной концентрации непрерывно в течение суток воздух со скоростью 10 л/ч протягивают через поглотительный прибор с пористым фильтром N 1, содержащий 6 мл поглотительного раствора. При наполнении прибора отмечают уровень жидкости, который периодически добавляют водой до первоначального.
Допустимо проводить отбор с перерывами в 2 — 4 ч в один и тот же прибор. Пробы должны быть проанализированы в течение 48 ч.
Ход анализа
В пробирку переносят 3 мл пробы, добавляют 2 мл фуксинформальдегидного реактива, содержимое пробирки перемешивают, через 20 мин. переносят в кювету шириной 1 см и измеряют оптическую плотность по сравнению с контролем с помощью фотоэлектроколориметра при длине волны 575 нм. Содержание вещества в анализируемом объеме определяют по заранее построенному калибровочному графику. Для построения графика готовят шкалу стандартов (табл. 8).
Таблица 8
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
|
Рабочий стандартный раствор, мл |
0 |
0,06 |
0,12 |
0,18 |
0,24 |
0,30 |
0,6 |
0,9 |
|
Поглотительный раствор, мл |
3 |
2,94 |
2,88 |
2,82 |
2,76 |
2,7 |
2,4 |
2,1 |
|
Содержание сернистого газа, мкг |
0 |
0,3 |
0,6 |
0,9 |
1,2 |
1,5 |
3,0 |
4,5 |
Все пробирки шкалы обрабатывают аналогично пробе, измеряют оптическую плотность и строят график. Для визуального определения строят шкалу стандартов (табл. 9).
Таблица 9
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
9 |
|
Рабочий стандартный раствор, мл |
0 |
0,02 |
0,06 |
0,1 |
0,2 |
0,4 |
0,8 |
1,2 |
1,6 |
2,0 |
|
Поглотительный раствор, мл |
2 |
1,98 |
1,94 |
1,9 |
1,8 |
1,6 |
1,2 |
0,8 |
0,4 |
— |
|
Содержание сернистого газа, мкг |
0 |
0,1 |
0,3 |
0,5 |
1,0 |
2,0 |
4,0 |
6,0 |
8,0 |
10 |
Для анализа берут 2 мл пробы.
Во все пробирки вливают по 1 мл фуксинформальдегидного реактива, растворы перемешивают. Определение проводят через 20 мин. после вливания реактива. Шкалу и пробу обрабатывают одновременно.
Расчет см. выше.
Примечания: а) При наличии окислов азота применяют поглотительный раствор, содержащий 0,06% аминосульфоновой кислоты или 0,08% аминосульфоната калия (NH2SO3K).
б) Раствор фуксина не должен иметь в диапазоне 600 — 750 нм в кювете шириной 1 см по сравнению с дистиллированной водой большую оптическую плотность, чем 0,05. При более высоких величинах оптической плотности раствор фуксина следует очистить. Раствор встряхивают 2 раза с бензолом, затем прибавляют на 1 л раствора до 20 г активированного угля, тщательно встряхивают в течение 1/2 — 1 ч и фильтруют через плотный аналитический фильтр («синяя лента»).
В случае необходимости раствор фильтруют через фильтр с активированным углем несколько раз до тех пор, пока оптическая плотность не достигнет нужных величин. Правильно приготовленный раствор окрашен в оранжево-желтый цвет, его оптическая плотность по сравнению с дистиллированной водой в кювете 1 см должна быть 0,02 — 0,03.
в) Для точной навески сульфита или пиросульфита при приготовлении исходного раствора следует установить точное содержание SO2 в применяемой соли путем йодометрического титрования.
г) В качестве поглотительного раствора можно применять 0,01 н. раствор едкого натра в 5% растворе глицерина.
Если же в качестве поглотительного раствора был взят 0,01 н. раствор едкого натра в 5% растворе глицерина, то сероводород, присутствующий в воздухе, удаляют с помощью твердого зернистого сорбента (зерна 4 — 5 мм) 10 мл пемзы, пропитанной насыщенным раствором ацетата свинца или сульфатом меди, высушенной при 105 — 110° и помещенной в стеклянную трубку. Последнюю присоединяют к входному отверстию поглотительного прибора в вертикальном положении [3].
ОПРЕДЕЛЕНИЕ СЕРНИСТОГО АНГИДРИДА С ПАРАРОЗАНИЛИНОМ [2]
Принцип определения
При взаимодействии сернистого газа с парарозанилином и формальдегидом в кислой среде раствор окрашивается в красно-фиолетовый цвет, по интенсивности которого определяют содержание сернистого газа.
Чувствительность определения — 0,5 мкг в анализируемом объеме раствора.
Двуокись азота мешает определению в концентрациях 4 мг/м3.
Реактивы
1. Поглотительный раствор 0,1 М раствор тетрахлормеркурата натрия: в мерной колбе емкостью 1 л, наполненной 800 мл воды, растворяют 11,7 г хлорида натрия и затем 27,2 г хлорида ртути х.ч. После полного растворения объем раствора в колбе доводят до метки водой.
Через несколько часов раствор фильтруют через плотный стеклянный фильтр и прозрачный раствор плотно закрывают; он устойчив в течение длительного времени. Раствор ядовит.
2. Стандартный раствор сернистого газа: исходный раствор с содержанием 4 мг/мл получают растворением 0,5936 г пиросульфита натрия х.ч. водой в мерной колбе емкостью 100 мл. Раствор применяют свежеприготовленный. Предварительно устанавливают содержание сернистого газа в 1 г соли.
Стандартный раствор А с содержанием 20 мкг/мл сернистого газа получают из 0,5 мл исходного раствора, который вливают в мерную колбу емкостью 100 мл и доводят раствор до метки поглотительным раствором. Раствор применяют всегда свежеприготовленным.
Рабочий стандартный раствор с содержанием 2 мкг/мл сернистого газа получают из стандартного раствора А при разбавлении его в 10 раз. Раствор применяют всегда свежеприготовленный.
3. Парарозанилин-хлоргидрат, 0,04% раствор: готовят в мерной колбе емкостью 500 мл, в которую наливают 400 мл воды и вносят 0,200 г парарозанилина-хлоргидрата х.ч. и растворяют. Через сутки прибавляют 30 мл соляной кислоты уд. веса 1,19, раствор доводят до метки водой и тщательно встряхивают. По истечении суток раствор готов. Хранят его в темном месте при нормальной температуре в плотно закрытой склянке. Раствор устойчив в течение месяца.
4. Формальдегид, 0,2% раствор: 2,5 мл 40% раствора формальдегида х.ч. разбавляют водой в мерной колбе емкостью 50 мл. Раствор устойчив в течение 2 нед.
Отбор пробы
а) Для определения разовой концентрации протягивают воздух со скоростью 2 л/мин. через поглотительный прибор с пористым фильтром N 1, содержащий 10 мл поглотительного раствора. Продолжительность отбора 20 — 30 мин.
б) Для определения среднесуточной концентрации воздух непрерывно в течение суток протягивают со скоростью 25 л/ч через поглотительный прибор с пористым фильтром N 1, в котором содержится 10 мл поглотительного раствора. При наполнении прибора отмечают уровень жидкости, который периодически добавляют водой до первоначального.
Допустимо проводить отбор с перерывами в 2 — 4 ч в один и тот же поглотительный прибор в течение 30 мин.
Отобранные пробы можно хранить в течение недели в темноте при комнатной температуре.
Ход анализа
В пробирку с притертой пробкой переносят 2 — 8 мл пробы, объем доводят до 8 мл поглотительным раствором, затем в пробирку вливают 1 мл раствора парарозанилина-хлоргидрата и 1 мл раствора формальдегида, содержимое пробирки встряхивают и через 20 мин. раствор переливают в кювету шириной 1 — 2 см и измеряют оптическую плотность по сравнению с контролем при длине волны 560 нм.
Окраска раствором устойчива в течение 30 мин.
Содержание сернистого газа в анализируемом объеме определяют по заранее построенному калибровочному графику. Для построения графика готовят шкалу стандартов (табл. 10).
Таблица 10
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
|
Рабочий стандартный раствор, мл |
0 |
0,25 |
0,5 |
1,0 |
2,0 |
4,0 |
6,0 |
8,0 |
|
Поглотительный раствор, мл |
8 |
7,75 |
7,5 |
7,0 |
6,0 |
4,0 |
2,0 |
0 |
|
Содержание сернистого газа, мкг |
0 |
0,5 |
1,0 |
2,0 |
4,0 |
8,0 |
12,0 |
16,0 |
Все пробирки шкалы обрабатывают аналогично пробе, измеряют оптическую плотность и строят график.
Расчет см. выше.
Шкала может быть использована для визуального определения.
Примечание. Если в воздухе присутствовал сероводород, то он, соединяясь с солями ртути, образует нерастворимый осадок сульфида ртути. Последний удаляют фильтрованием или центрифугированием.
ОПРЕДЕЛЕНИЕ СЕРНИСТОГО АНГИДРИДА С ХЛОРИДОМ БАРИЯ [6]
Принцип определения
Сернистый газ окисляется хлоратом калия до серной кислоты, которая образует с хлоридом бария нерастворимый сульфат бария. Интенсивность помутнения пропорциональна концентрации сернистого газа.
Чувствительность определения — 3 мкг в анализируемом объеме раствора. Серная кислота и растворимые сульфаты мешают определению.
Реактивы
1. Поглотительный раствор. 4% раствор хлората калия: 4 г перекристаллизованного хлората калия растворяют в 100 мл дважды дистиллированной воды, в которой не содержится сульфатов.
2. Рабочий стандартный раствор сернистого газа готовят растворением 0,0272 г сульфата калия в воде, в мерной колбе емкостью 100 мл; 1 мл раствора соответствует 100 мкг сернистого газа.
3. Соляная кислота, 0,3% раствор.
4. Хлорид бария, 5% раствор.
5. Спирт этиловый, 96°.
6. Этиленгликоль.
7. Составной реактив. Готовят смешением трех объемов этиленгликоля с одним объемом 5% раствора хлорида бария и тремя объемами этилового спирта. Величину pH смеси доводят до 2,5 — 2,8 (проверяют на pH-метре). Составной реактив готовят за 2 дня до употребления. Годен к применению в течение 2 мес.
Отбор пробы
а) Для определения разовой концентрации воздух со скоростью 2 л/мин. протягивают через поглотительный прибор с пористым фильтром N 1, содержащий 6 мл поглотительного раствора. Продолжительность отбора 20 — 30 мин.
б) Для определения среднесуточной концентрации непрерывно в течение суток протягивают воздух со скоростью 0, 25 л/мин. через поглотительный прибор, содержащий 6 мл поглотительного раствора. При наполнении прибора отмечают уровень жидкости, который периодически доводят до первоначального. Допустимо проводить отбор 6 — 12 раз с перерывами в 2 — 4 ч в один и тот же поглотительный прибор.
Ход анализа
Содержимое поглотительного прибора доводят до первоначального объема бидистиллятом. 3 мл раствора помещают в пробирку, добавляют 2 мл составного реактива и осторожно перемешивают, избегая образования пузырьков. Через 5 мин. определяют интенсивность помутнения раствора в кюветах с толщиной слоя 1 см, при длине волны 410 или 540 нм по сравнению с контролем. Содержание сернистого газа в анализируемом объеме раствора определяют по заранее построенному калибровочному графику.
Для построения калибровочного графика готовят шкалу стандартов (табл. 11).
Таблица 11
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
|
Рабочий стандартный раствор, мл |
0 |
0,03 |
0,05 |
0,1 |
0,2 |
0,4 |
0,6 |
|
Поглотительный раствор, мл |
3 |
2,97 |
2,95 |
2,9 |
2,8 |
2,6 |
2,4 |
|
Содержание сернистого газа, мкг |
0 |
3 |
5 |
10 |
20 |
40 |
60 |
Все пробирки шкалы и пробы обрабатывают аналогично пробе, измеряют оптические плотности и строят график.
Шкала может быть использована для визуального определения.
Расчет см. выше.
ОПРЕДЕЛЕНИЕ СЕРНОЙ КИСЛОТЫ С ХЛОРИДОМ БАРИЯ [7]
Принцип определения
Серная кислота, взаимодействуя с хлоридом бария, образует сульфат бария, нерастворимый в воде. Интенсивность помутнения раствора пропорциональна количеству серной кислоты.
Чувствительность определения — 3 мкг серной кислоты в анализируемом объеме раствора. Определению мешают сульфаты, сернистый газ определению не мешает.
Реактивы
1. Исходный стандартный раствор готовят растворением 0,1776 г перекристаллизованного сернокислого калия в мерной колбе емкостью 100 мл в бидистиллированной воде. 1 мл раствора соответствует 1 мг серной кислоты.
Рабочий стандартный раствор с содержанием 100 мкг/мл готовят соответствующим разбавлением.
2. Реактивы — см. «Определение сернистого газа с хлоридом бария».
Отбор пробы
а) Для определения разовой концентрации исследуемый воздух со скоростью 10 л/мин. протягивают через фильтр АФА, укрепленный в патрон. Продолжительность отбора 20 мин.
б) Для определения среднесуточной концентрации исследуемый воздух со скоростью 10 л/мин. протягивают через фильтр АФА непрерывно в течение суток.
Допустимо среднесуточный отбор проводить на один и тот же фильтр 6 — 12 раз с перерывами в 2 — 4 ч.
Ход анализа
Вынимают фильтр из патрона, обрезают края и помещают в воронку с пористой пластинкой N 1. Воронку вставляют в пробирку с меткой на 10 мл, имеющую боковой отросток, который присоединен к водоструйному насосу. Наносят на фильтр 1 мл этанола и промывают его 4 — 5 раз горячим бидистиллятом. Доводят объем до 10 мл. Для контроля таким же образом промывают неиспользованный фильтр.
К 3 мл пробы добавляют 2 мл составного реактива и осторожно перемешивают, избегая образования воздушных пузырьков. Через 5 мин. определяют интенсивность помутнения раствора в кюветах с толщиной слоя 1 см, при длине волны 365 нм по сравнению с контролем. Если в пробе ожидается малое содержание серной кислоты, то пробу упаривают до объема 5 мл на кипящей водяной бане, для анализа берут также 3 мл. Содержание серной кислоты устанавливают по калибровочному графику. Для построения калибровочного графика готовят шкалу стандартов (табл. 12).
Таблица 12
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
|
Рабочий стандартный раствор, мл |
0 |
0,03 |
0,05 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
|
Поглотительный раствор, мл |
3 |
2,97 |
2,95 |
2,9 |
2,8 |
2,6 |
2,4 |
2,2 |
|
Содержание серной кислоты, мкг |
0 |
3 |
5 |
10 |
20 |
40 |
60 |
80 |
Все пробирки шкалы и пробы обрабатывают аналогично пробе, измеряют оптическую плотность и строят калибровочный график.
Шкала может быть использована для визуального определения.
Расчет см. выше.
ОПРЕДЕЛЕНИЕ СЕРНИСТОГО ГАЗА И СЕРНОЙ КИСЛОТЫ [6]
Принцип определения
Аэрозоль серной кислоты отбирают на фильтр АФА. Сернистый газ фильтром не задерживается, а поглощается в поглотительном приборе Рыхтера, наполненном поглотительным раствором.
Серную кислоту определяют нефелометрическим методом, сернистый газ — колориметрическим или нефелометрическим методами.
Расчет см. выше.
Отбор пробы
а) Для определения разовой концентрации воздух со скоростью 3 — 4 л/мин. протягивают через фильтр АФА, помещенный в патрон, к которому присоединен поглотительный прибор Рыхтера, содержащий 6 мл поглотительного раствора. Продолжительность отбора 20 — 30 мин.
б) Для определения среднесуточной концентрации воздух со скоростью 3 — 4 л/мин. протягивают через аналогичную систему в течение суток. Допустимо среднесуточный отбор проводить в один и тот же фильтр 6 — 12 раз с перерывами в 2 — 4 ч.
Ход анализа см. выше.
ОПРЕДЕЛЕНИЕ СЕРНОЙ КИСЛОТЫ ПО ИОНУ ВОДОРОДА [8]
Принцип определения
Серная кислота окрашивает раствор индикатора водного голубого в синий цвет. Интенсивность окраски пропорциональна концентрации водородных ионов серной кислоты.
Чувствительность определения — 0,01 мкг водородных ионов в анализируемом объеме раствора.
Определению мешают аэрозоли щелочей. Сернистый газ определению не мешает.
Реактивы
1. Дистиллированная вода, дважды перегнанная, pH 5,4 — 6,6.
2. Исходный стандартный раствор серной кислоты. 0,005 н. раствор серной кислоты готовят соответствующим разбавлением 0,1 н. раствора серной кислоты. 1 мл раствора содержит 5 мкг водородных ионов. Рабочий стандартный раствор готовят разбавлением исходного раствора в 10 раз. 1 мл рабочего раствора содержит 0,5 мкг ионов водорода.
3. 0,02% раствор индикатора водного голубого в воде.
Отбор пробы
а) Для определения разовой концентрации исследуемый воздух со скоростью 10 л/мин. протягивают через фильтр АФА, помещенный в патрон. Продолжительность отбора 20 — 30 мин.
б) Для определения среднесуточной концентрации воздух со скоростью 5 л/мин. протягивают через фильтр непрерывно в течение суток. Допустимо отбор проводить в один и тот же фильтр 6 — 12 раз с перерывами в 2 — 4 ч.
Ход анализа
Фильтр вынимают из патрона, срезают края и помещают в воронку с пористой пластинкой N 1. Воронку вставляют в пробирку с меткой на 10 мл, имеющую боковой отросток, который присоединен к водоструйному насосу. Наливают на фильтр 1 мл этанола и промывают фильтр 4 — 5 раз горячей водой до общего объема 10 мл. Так же промывают неиспользованный фильтр для контроля.
Для анализа берут 5 мл промывной жидкости, добавляют 0,5 мл 0,02% раствора водного голубого и взбалтывают содержимое. Через 30 мин. измеряют интенсивность окраски раствора в кюветах с толщиной слоя 1 см при длине волны 592 нм по сравнению с контролем.
Содержание серной кислоты устанавливают по калибровочному графику. Для построения калибровочного графика готовят шкалу стандартов (табл. 13).
Таблица 13
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
|
Рабочий стандартный раствор, мл |
0 |
0,02 |
0,05 |
0,1 |
0,2 |
0,3 |
0,4 |
0,5 |
|
Поглотительный раствор, мл |
5 |
4,98 |
4,95 |
4,9 |
4,8 |
4,7 |
4,7 |
4,5 |
|
Содержание серной кислоты, мкг |
0 |
0,01 |
0,025 |
0,05 |
0,1 |
0,15 |
0,2 |
0,25 |
Все пробирки шкалы обрабатывают аналогично пробе, измеряют оптическую плотность и строят график. Шкала может быть использована для визуального определения.
Расчет см. выше.
Примечание. Для выражения результатов анализа по молекуле серной кислоты надо провести пересчет. Коэффициент пересчета 49,05.
СЕРОВОДОРОД
Бесцветный газ, при большом разбавлении имеет неприятный запах, плотность по отношению к воздуху 1,1906, тяжелее воздуха. Вес 1 л газа 1,5392 г, он легко конденсируется в бесцветную жидкость. Сероводород растворим в воде. Один объем воды при 0 °C может поглотить 4,65, при 10 °C — 3,44 и при 20 °C — 2,61 объема сероводорода; он хорошо растворим в спирте: один объем спирта растворяет 11,8 объема при 10 °C. Сероводород является энергичным восстановителем, водный раствор его имеет слабокислую реакцию.
Сероводород — сильный нервный яд, небольшие концентрации которого действуют на слизистые оболочки дыхательных путей и глаз, вызывают головную боль, светобоязнь и боль в глазницах.
Предельно допустимые концентрации: максимальная разовая и среднесуточная 0,008 мг/м3.
ОПРЕДЕЛЕНИЕ СЕРОВОДОРОДА С ПАРААМИНОДИМЕТИЛАНИЛИНОМ [4]
Принцип определения
При взаимодействии сероводорода с парааминодиметиланилином в кислой среде в присутствии трехвалентного железа образуется метиленовая синь. По интенсивности окраски раствора определяют содержание сероводорода.
Чувствительность определения 1 мкг в анализируемом объеме раствора.
Хлор, окислы азота мешают определению в концентрациях, в 5 раз превышающих сероводород, сернистый газ — в концентрациях, превышающих в 20 раз.
Реактивы
1а. Поглотительный раствор: 2,15 г сернокислого кадмия (CdSO4·8H2O) растворяют в 150 мл воды в колбе емкостью 500 мл и затем добавляют в колбу емкостью 100 мл 0,1 н. раствора едкого натра (постепенно, при взбалтывании), при этом образуется осадок гидроокиси кадмия. Раствор в колбе доводят водой до метки. При применении раствор хорошо взбалтывают. Эта суспензия применяется в качестве поглотительного раствора.
1б. В качестве поглотительного раствора можно применять 2% раствор ацетата цинка, подкисленного ледяной уксусной кислотой, 0,2 мл кислоты на 100 мл воды.
2. Стандартный раствор сероводорода. Исходный раствор готовят насыщением сероводородом 0,1 н. раствора едкого натра, приготовленного на дважды перегнанной воде.
Содержание сероводорода в 1 мл раствора устанавливают йодометрически. В коническую колбу вносят 10 мл приготовленной сероводородной воды, в которую предварительно было налито 20 мл 0,1 н. раствора йода, подкисленного 2 мл 5 н. соляной кислоты. Избыток йода, не связанный с сероводородом, оттитровывают 0,1 н. раствором тиосульфата в присутствии 0,5% раствора крахмала. 1 мл 0,1 н. раствора тиосульфата соответствуют 1,7043 мг сероводорода.
Для приготовления стандартного раствора А с содержанием 50 мкг/мл в мерную колбу емкостью 100 мл вносят такое количество исходного раствора, в котором содержится 5 мг сероводорода, и колбу доливают дважды перегнанной водой до метки.
Содержание сероводорода в 1 мл устанавливают йодометрически, титруя раствор 0,005 н. тиосульфатом, 1 мл которого соответствует 85,20 мкг сероводорода.
Рабочий стандартный раствор с содержанием 2 мкг/мл готовят из раствора А в мерной колбе емкостью 100 мл, в которую вливают количество раствора А, содержащее 200 мкг сероводорода. Перед приготовлением рабочего стандартного раствора раствор А необходимо титровать.
Рабочий стандартный раствор устойчив в течение 40 — 50 мин.
3. Парааминодиметиланилин сульфат, 0,1% раствор: 0,05 г растворяют в колбе емкостью 50 мл в 25 мл соляной кислоты, уд. вес 1,19, и добавляют водой до метки. Сохраняется в течение 2 нед. в холодном темном месте.
4. Хлорное железо, 0,12% раствор. 0,12 г трехвалентного хлорного железа растворяют в 10 мл соляной кислоты, уд. вес 1,19, и объем раствора доводят до 100 мл водой.
5. Едкий натр, 0,1 н. раствор.
6. Тиосульфат, 0,1 н. раствор. Тиосульфат 0,005 н. раствор готовят перед применением из 0,1 н. раствора.
7. Йод, 0,1 н. раствор.
8. Сорбент для задержания сернистого ангидрида. Пемзу с размером частиц 3 — 5 мм пропитывают насыщенным раствором ацетата натрия, высушивают в сушильном шкафу при 110 — 120 °C и наполняют стеклянный патрон (диаметр 2 см), в который насыпают 8 — 10 см3 сорбента. Патрон соединяют с входным отверстием поглотительного прибора, помещая его в вертикальном положении.
Отбор пробы
а) Для определения разовой концентрации протягивают воздух со скоростью 2 л/мин. через два поглотительных прибора с пористым фильтром N 1, содержащие по 1 мл поглотительного раствора и 4 мл воды. Продолжительность отбора 20 — 30 мин. Поглотительный прибор должен быть защищен от света.
б) Для определения среднесуточной концентрации непрерывно в течение суток воздух со скоростью 25 л/ч протягивают через два поглотительных прибора с пористым фильтром N 1, содержащих по 1 мл поглотительного раствора и 7 мл воды.
При наполнении прибора отмечают уровень жидкости, который периодически добавляют водой до первоначального.
Прибор защищают от света.
Допустимо проводить отбор 6 — 12 раз с перерывами в 4 — 2 ч в один и тот же прибор.
Ход анализа
Каждый поглотительный прибор анализируют отдельно. Раствор из поглотительного прибора переносят в мерную колбу емкостью 10 мл или в пробирку с меткой 10 мл и с притертой пробкой, добавляют 0,5 мл раствора парааминодиметиланилина, 0, 5 мл раствора хлорного железа и через 10 мин. доливают водой до метки. Через 10 мин. раствор переводят в кювету шириной 2 см и измеряют оптическую плотность по сравнению с контролем, который готовят одновременно и аналогично пробе при длине волны 665 нм.
Содержание сероводорода в анализируемом объеме определяют по предварительно построенному калибровочному графику.
Для построения калибровочного графика готовят шкалу стандартов в колбах емкостью 10 мл (табл. 14).
Таблица 14
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
|
Рабочий стандартный раствор, мл |
0 |
0,5 |
1,0 |
1,5 |
2,0 |
2,5 |
|
Поглотительный раствор, мл |
По 1 мл в каждую колбу |
|||||
|
Содержание сероводорода, мкг |
0 |
1 |
2 |
3 |
4 |
5 |
Объем растворов доводят до 10 мл и далее обрабатывают аналогично пробе.
Измеряют оптическую плотность и строят калибровочный график.
Для визуального определения готовят шкалу согласно табл. 15.
Таблица 15
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
|
Рабочий стандартный раствор, мл |
0 |
0,15 |
0,25 |
0,5 |
1 |
1,5 |
2 |
|
Поглотительный раствор, мл |
По 1 мл |
||||||
|
Вода, мл |
4 |
3,85 |
3,75 |
3,5 |
3 |
2,5 |
2 |
|
Содержание сероводорода, мкг |
0 |
0,3 |
0,5 |
1 |
2 |
3 |
4 |
Для анализа берут всю пробу, переносят в колориметрические пробирки, вносят по 0,25 мл раствора парааминодиметиланилина и по 0,25 мл раствора хлорного железа. Через 10 мин. проводят определение.
Расчет см. выше.
Примечания: 1. Для анализа, кроме сульфата парааминодиметиланилина, можно использовать и хлоргидрат, приготовленный следующим образом. Сначала готовят 0,5% раствор метилового оранжевого. Для этого навеску 0,5 г растворяют в воде в мерной колбе емкостью 100 мл при нагревании (50 — 60 °C). После охлаждения до комнатной температуры раствор доводят водой до метки. Перед применением раствор слегка нагревают для растворения выпавших кристаллов.
К 10 мл 0,5% раствора метилового оранжевого прибавляют 10 мл концентрированной соляной кислоты, перемешивают и добавляют для восстановления 5 г порошкообразного металлического цинка, который прибавляют небольшими порциями до обесцвечивания раствора. Последнее происходит через 15 — 20 мин. Затем цинк быстро отфильтровывают через вату, промывают 10 мл воды, к прозрачному раствору прибавляют еще 15 мл концентрированной соляной кислоты и доводят объем водой до 50 мл.
Реактив хранят в темном и холодном месте, его можно использовать до тех пор, пока он бесцветный или бледно-желтого цвета. Перед употреблением реактив следует проверять на стандартных растворах.
2. Отобранные пробы хранят в темном и холодном месте, их анализ рекомендуется производить в течение суток.
ОПРЕДЕЛЕНИЕ СЕРОВОДОРОДА С НИТРАТОМ СЕРЕБРА [5]
Принцип определения
При взаимодействии сероводорода с нитратом серебра образуется коллоидальный раствор сульфида серебра, окрашенный в желто-коричневый цвет. По интенсивности окраски раствора определяют содержание сероводорода.
Чувствительность определения 1 мкг в анализируемом объеме раствора.
Мешают определению галоиды.
Реактивы
1. Поглотительный раствор готовят из ортоарсенита натрия или мышьяковистого ангидрида.
В первом случае растворяют 2 г арсенита натрия в 100 мл 5% раствора карбоната аммония и объем раствора доводят до 1 л водой.
Во втором случае 2 г мышьяковистого ангидрида и 5 г карбоната аммония вносят в небольшой объем воды, подогревают до растворения ангидрида и доводят объем раствора до 1 л водой.
2. Стандартный раствор сероводорода. Исходный раствор с содержанием 100 мкг/мл готовят следующим образом: вливают 3 мл 0,1 н. раствора тиосульфата в мерную колбу емкостью 100 мл и добавляют до метки 0,5% раствором карбоната аммония.
Рабочий стандартный раствор с содержанием 10 мкг/мл получают разбавлением исходного раствора в 10 раз поглотительным раствором. Готовят перед анализом.
3. Нитрат серебра (1% раствор): 1 г нитрата серебра растворяют в 80 мл воды и осторожно добавляют 10 мл концентрированной серной кислоты. После охлаждения раствор доливают водой до 100 мл.
4. Карбонат аммония (0,5% раствор).
5. Крахмал (1% раствор).
Отбор пробы
а) Для определения разовой концентрации воздух со скоростью 4 л/мин. протягивают через поглотительный прибор Рыхтера, содержащий 6 мл поглотительного раствора.
Продолжительность отбора 30 мин.
б) Для определения среднесуточной концентрации воздух непрерывно в течение суток со скоростью 25 л/ч протягивают через поглотительный прибор с пористым фильтром N 1, содержащий 6 мл поглотительного раствора. При наполнении прибора отмечают уровень жидкости, который периодически добавляют водой до первоначального уровня.
Допустимо проводить отбор с перерывами в 2 — 4 ч в один и тот же прибор 6 — 12 раз.
Ход анализа
В колориметрическую пробирку переносят 5 мл пробы. Одновременно готовят шкалу стандартов (табл. 16).
Таблица 16
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
|
Рабочий стандартный раствор, мл |
0 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
|
Поглотительный раствор, мл |
5 |
4,9 |
4,8 |
4,6 |
4,4 |
4,2 |
4,0 |
|
Содержание сероводорода, мкг |
0 |
1 |
2 |
4 |
6 |
8 |
10 |
Во все пробирки шкалы и проб наливают по 0,2 мл крахмала и по 1 мл нитрата серебра. Содержимое пробирок встряхивают и через 5 мин. производят сравнения интенсивности окраски проб со шкалой стандартов.
Расчет см. выше.
Примечание. Определение можно проводить фотометрически при длине волны 346 нм в кювете с толщиной слоя 1 — 2 см. Чувствительность определения 5 мкг в анализируемом объеме раствора.
СЕРОУГЛЕРОД
Бесцветная жидкость, в чистом виде имеет приятный запах, но при наличии примесей обычно обладает неприятным запахом. Удельный вес при 25 °C 1,263, кипит при 46,3 °C. Пары чрезвычайно легко воспламеняются, они тяжелее воздуха в 2,6 раза. Сероуглерод нерастворим в воде, но хорошо растворяется в ряде органических растворителей; он является хорошим растворителем для жиров, масел, смол, каучука, серы, фосфора, йода и некоторых других трудно растворимых веществ.
Сероуглерод токсичен, действует на центральную нервную систему, поражает и внутренние органы.
Предельно допустимые концентрации: максимальная разовая и среднесуточная 0,008 мг/м3.
ОПРЕДЕЛЕНИЕ СЕРОУГЛЕРОДА С ДИЭТИЛАМИНОМ [2]
Принцип определения
Сероуглерод реагирует с диэтиламином, образуя диэтилдитиокарбамат, который при наличии ионов двухвалентной меди образует желто-коричневую окраску вследствие образования комплексной соли. Интенсивность окраски раствора пропорциональна количеству сероуглерода.
Чувствительность: 1 мкг CS2 в определяемом объеме.
Определению мешает сероводород.
Влияние сероводорода устраняют применением предохранительного фильтра. Меркаптаны, тиофен, диметилсульфид определению не мешают.
Реактивы
1. Поглотительный раствор: 0,1 г ацетата меди х.ч. (CH3COO)·CuH2O растворяют в 5 мл воды и 10 мл дважды перегнанного триэтаноламина, прибавляют 5 мл чистого диэтиламина (или 15 мл 33% раствора) и доводят этанолом до 1 л. Можно пользоваться также этанолом, содержащим 1% бензина. Если приготовленный поглотительный раствор слегка опалесцирует, ему дают перед применением отстояться в течение 2 — 3 дней. Реактив в хорошо закрытой бутылке устойчив в течение нескольких недель.
2. Стандартный раствор сероуглерода. Исходный раствор готовят в мерной колбе емкостью 50 мл, в которую вливают 10 — 15 мл этанола. Колбу взвешивают и вносят в нее 2 — 3 капли свежеперегнанного сероуглерода и вновь взвешивают. Объем жидкости в колбе доводят до метки этанолом и рассчитывают количество сероуглерода в 1 мл.
Рабочий стандартный раствор с содержанием сероуглерода 2 мкг/мл получают в мерной колбе емкостью 50 мл, в которую предварительно вливают 15 — 20 мл этанола, а затем вносят такое количество исходного раствора, которое содержит 100 мкг сероуглерода, и раствор доводят до метки этанолом.
3. Вата для улавливания H2S [3].
В 50 мл воды, подкисленной несколькими каплями уксусной кислоты, растворяют 10 г ацетата свинца, прибавляют 10 мл глицерина и доводят водой до 100 мл. Хлопчатобумажную вату, пропитанную этим раствором, выжимают, высушивают при комнатной температуре и хранят в банке с притертой пробкой.
Отбор пробы
а) Для определения разовой концентрации исследуемый воздух со скоростью 0,5 л/мин. протягивают через поглотительный прибор, наполненный 5 мл поглотительного раствора. К поглотителю присоединена стеклянная трубочка, наполненная ватой для улавливания сероводорода.
Продолжительность отбора 20 — 30 мин. при охлаждении.
б) Для определения среднесуточной концентрации непрерывно в течение суток воздух со скоростью 7,5 л/ч протягивают через поглотительный прибор, содержащий 6 мл поглотительного раствора. При наполнении прибора отмечают уровень жидкости, который периодически добавляют этанолом до первоначального 33% раствором диэтиламина.
Допустимо проводить отбор 6 — 12 раз с перерывами в 2 — 4 ч в один и тот же поглотительный прибор.
Ход анализа
Содержимое поглотительного прибора переносят в пробирку емкостью 10 мл и доводят до метки поглотительным раствором. Интенсивность окраски измеряют через 15 мин. с помощью фотометра в кювете шириной от 1 до 5 см при длине волны 465 нм по сравнению с контролем (поглотительным раствором). Окраска устойчива в течение суток.
Содержание сероуглерода в анализируемом объеме определяют по предварительно построенному калибровочному графику. Для построения калибровочного графика готовят шкалу стандартов (табл. 17).
Таблица 17
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
9 |
10 |
|
Рабочий стандартный раствор, мл |
0 |
0,5 |
1,0 |
2,0 |
3,0 |
4,0 |
5,0 |
6,0 |
7,0 |
8,0 |
10 |
|
Содержание сероуглерода, мкг |
0 |
1,0 |
2,0 |
4,0 |
6,0 |
8,0 |
10,0 |
12 |
14 |
16 |
20 |
Объем каждой пробирки доводят до метки поглотительным раствором. Измеряют оптическую плотность и строят калибровочный график в координатах концентрация — оптическая плотность. Шкала может быть использована для визуального определения.
Расчет см. выше.
ЛИТЕРАТУРА
1. Алексеев М.В., Самородина Р.Я. Колориметрическое определение содержания сернистого газа в атмосферном воздухе. Гиг. и сан., 1953, 10.
2. Определение сернистого газа по реакции с фуксином. Определение сернистого газа по реакции с парарозанилином. Определение сероуглерода. В сб.: СЭВ. Унифицированные методы определения атмосферных загрязнений. Ч. 1. М., 1970.
3. Павленко А.А., Кузьмина Т.А. Предохранительный патрон для задержания сероводорода, сернистого газа. Труды Главной геофизической обсерватории. Л., 1968, в. 234.
4. Пауль И.И., Виноградова В.А. Определение малых количеств сероводорода в воздухе. Гигиена труда, 1960, 2.
5. Полежаев Н.Г. К методике определения малых количеств сероводорода в воздухе. Гигиена труда и техника безопасности, 1937, 6.
6. Чепкевичене Е.С. Разработка методов определения серной кислоты и раздельного определения серной кислоты, сернистого ангидрида и сульфатов в воздухе при гигиенических исследованиях. Автореф. дисс. канд. М., 1972.
7. Чепкевичене Е.С. К вопросу определения аэрозоля серной кислоты в атмосферном воздухе. Вопросы эпидемиологии и гигиены в Литовской ССР. Вильнюс, 1969.
8. Чепкевичене Е.С. Определение серной кислоты по водородному катиону. Вопросы эпидемиологии и гигиены в Литовской ССР. Вильнюс, 1971.
Глава IV
СОЕДИНЕНИЯ АЗОТА
АММИАК
Бесцветный газ с резким запахом, хорошо растворимый в воде, в спирте растворяется 13,2% при 20 °C.
В продаже бывает в виде водных растворов с содержанием 28 — 29% аммиака (удельный вес 0,90 при 25 °C). Плотность аммиака по отношению к воздуху 0,59.
Температура плавления 77,7 °C, температура кипения 33,35 °C. Вес 1 л газообразного аммиака при нормальных условиях 0,771 г.
Аммиак оказывает раздражающее действие на верхние дыхательные пути, а также на слизистые оболочки глаз.
Предельно допустимые концентрации аммиака; максимальная разовая 0,20 мг/м3, среднесуточная 0,20 мг/м3.
ОПРЕДЕЛЕНИЕ АММИАКА С РЕАКТИВОМ НЕССЛЕРА [2]
Принцип определения
При взаимодействии аммиака с реактивом Несслера образуется соединение, окрашенное в желто-бурый цвет. Интенсивность окраски пропорциональна количеству иона аммония.
Чувствительность определения 2 мкг аммиака в анализируемом объеме раствора.
Определению мешают аммонийные соли, некоторые амины алифатического ряда, формальдегид с 0,2 мг/м3, сероводород с 0,04 мг/м3, хлор с 0,2 мг/м3. Окислы азота определению не мешают.
Реактивы
Все реактивы готовят на дистиллированной воде, не дающей положительной реакции на ион ![]()
1. Поглотительный раствор: серная кислота 0,01 н. раствор.
2. Стандартный раствор.
Исходный раствор с содержанием 100 мкг/мл аммиака готовят растворением 0,3140 г хлорида аммония в 100 мл дистиллированной воды.
Раствор устойчив в течение 1 — 2 мес.
Рабочий стандартный раствор, содержащий 10 мкг/мл аммиака, готовят разведением исходного раствора 100 раз водой. Раствор готовят перед анализом.
3. Реактив Несслера.
а) 17 г хлорида ртути растворяют в 300 мл воды, в другой колбе растворяют 35 г йодида калия в 100 мл воды. Ко второму раствору приливают первый до образования красного нерастворимого осадка, затем добавляют 600 мл 20% раствора едкого натра и оставшееся количество раствора хлорида ртути, раствору дают отстояться в темном месте, после чего осторожно сифонируют в темную склянку и закрывают резиновой пробкой.
При отстаивании в реактиве может образоваться осадок, но если жидкость над осадком слегка желтоватого цвета, прозрачный раствор пригоден к употреблению.
б) 10 г йодида ртути растирают в ступке с небольшими количествами воды (около 10 мл), переносят эту взвесь в склянку из темного стекла, ступку ополаскивают небольшим количеством воды, сливая ее в ту же склянку, и прибавляют 5 г йодида калия и 40 г едкого натра, растворенного в 60 — 70 мл дистиллированной воды. Реактив оставляют на 1 — 2 дня, после чего осторожно сифонируют прозрачный раствор в склянку из темного стекла и закрывают резиновой пробкой.
4. Дистиллированная вода, не дающая положительной реакции на ион ![]() Для ее получения к 1 л дистиллированной воды прибавляют 5 мл 10% серной кислоты и перегоняют ее. Первые порции погона (100 — 200 мл) отбрасывают. Следующий погон проверяют с реактивом Несслера на содержание иона
Для ее получения к 1 л дистиллированной воды прибавляют 5 мл 10% серной кислоты и перегоняют ее. Первые порции погона (100 — 200 мл) отбрасывают. Следующий погон проверяют с реактивом Несслера на содержание иона ![]() При отрицательной реакции воду собирают и хранят в закрытой бутыли.
При отрицательной реакции воду собирают и хранят в закрытой бутыли.
Безаммиачную воду можно получать также при пропускании дистиллированной воды со скоростью 5 мл в мин. через колонку с катионитами КУ-2 или СБС. Предварительно проводят подготовку катионита. С этой целью катионит помещают в стакан и заливают дистиллированной водой. На следующий день катионит многократно обрабатывают 5% раствором соляной кислоты для освобождения его от железа, проверяют присутствие его качественной реакцией с роданистым аммонием. Далее катионит переносят в бюретку емкостью 50 — 100 мл, смывая его дистиллированной водой. В нижнюю часть бюретки предварительно помещают слой стеклянной ваты толщиной 1 — 2 см.
Соляную кислоту пропускают через катионит медленно, до отрицательной реакции на железо. Подготовка катионита заканчивается промыванием его дистиллированной водой до нейтральной реакции.
Катионит следует хранить под слоем дистиллированной воды.
Отбор пробы
а) Для определения разовой концентрации воздух со скоростью 1 л/мин. протягивают через 2 поглотительных прибора с пористой пластинкой N 1, содержащих по 6 мл 0,01 н. раствора серной кислоты. Продолжительность отбора 20 — 30 мин.
б) Для определения среднесуточной концентрации воздух со скоростью 0,10 — 0,2 л/мин. протягивают через два поглотительных прибора с пористой пластинкой N 1, содержащих по 6 мл 0,01 н. раствора серной кислоты, отмечая уровень жидкости. Отбор проводят непрерывно в течение суток, периодически добавляя воду до первоначального уровня.
Допустимо проводить отбор 6 — 12 раз с перерывами 2 — 4 ч в одни и те же поглотительные приборы.
Ход анализа
Из каждого поглотительного прибора 5 мл пробы вносят в колориметрические пробирки, приливают по 0,5 мл реактива Несслера, взбалтывают и через 5 — 10 мин. фотометрируют в кюветах с толщиной слоя 1 — 2 см при длине волны 450 нм по сравнению с контролем, который готовят одновременно и аналогично пробам. Содержание аммиака в анализируемом объеме определяют по предварительно построенному калибровочному графику. Для построения калибровочного графика готовят шкалу стандартов (табл. 18).
Таблица 18
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
|
Рабочий стандартный раствор, мл |
0 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
2 |
|
Поглотительный раствор, мл |
5 |
4,8 |
4,6 |
4,6 |
4,2 |
4,0 |
3,0 |
|
Содержание аммиака, мкг |
0 |
2 |
4 |
6 |
8 |
10 |
20 |
Все пробирки шкалы обрабатывают аналогично пробам, измеряют оптическую плотность и строят график. Шкалой стандартов можно пользоваться для визуального определения, ее готовят в колориметрических пробирках одновременно с пробами.
Расчет см. выше.
Примечание. Реактив Несслера следует вливать быстро, тогда устраняется опалесценция раствора.
ОПРЕДЕЛЕНИЕ АММИАКА С НАТРИЕВОСАЛИЦИЛАТНЫМ РЕАКТИВОМ [2]
Принцип определения
Метод основан на образовании соединения, окрашенного в зеленовато-синий цвет при взаимодействии аммиака с натриевосалицилатным реактивом в присутствии гипохлорита натрия.
Чувствительность определения 0,1 мкг аммиака в анализируемом объеме раствора.
Сернистый газ в концентрации 3 мг/м3, сероводород — 0,2 мг/куб. м, сероуглерод — 0,7 мг/куб. м, фенол — 0,1 мг/м3 и альдегиды не мешают определению.
Реактивы
1. Поглотительный раствор: 0,02 н. раствор серной кислоты.
2. Исходный стандартный раствор с содержанием 100 мкг/мл аммиака готовят растворением 0,0314 г хлорида аммония в дважды перегнанной воде, не содержащей аммиака.
Рабочий стандартный раствор с содержанием 1 мкг/мл аммиака готовят разведением исходного раствора поглотительным раствором.
3. Едкий натр.
4. Хлорная вода (0,5 — 0,6% раствор).
5. Гипохлоритный реактив. 16 г едкого натра растворяют в 100 мл 0,5 — 0,6% хлорной воды в мерной колбе емкостью 100 мл.
6. Салицилат натрия.
7. Нитропруссид натрия.
8. Натриевосалицилатный реактив. К 45 г салицилата натрия прибавляют 50 мг нитропруссида натрия, предварительно хорошо растертого в ступке с 73 мл дважды перегнанной воды. Реактив сохраняют в темном полиэтиленовом сосуде, в темноте. При комнатной температуре реактив устойчив в течение 5 мес.
Отбор пробы
а) Для определения разовой концентрации воздух со скоростью 1 л/мин. протягивают через поглотительный прибор с пористым фильтром N 1, содержащий 6 мл поглотительного раствора. Продолжительность отбора 20 — 30 мин.
б) Для определения среднесуточной концентрации воздух со скоростью 0,5 л/мин. непрерывно в течение суток протягивают через поглотительный прибор с пористым фильтром N 1, содержащий 6 мл поглотительного раствора. Допустимо проводить отбор 6 — 12 раз с перерывом в 4 — 2 ч в один и тот же поглотительный прибор.
Ход анализа
Содержимое поглотительного прибора доводят до объема 6 мл поглотительным раствором, 5 мл раствора берут на анализ, добавляют 0,5 мл натриево-салицилатного реактива, взбалтывают, приливают 0,5 мл гипохлоритного реактива и снова взбалтывают. Через 2 1/2 — 3 ч фотометрируют в кюветах с толщиной слоя 1 см при длине волны 597 нм по сравнению с контролем, который готовят одновременно и аналогично пробе.
Содержание аммиака в анализируемом объеме определяют по градуировочному графику, который предварительно строят по шкале стандартов (табл. 19).
Таблица 19
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
|
Рабочий стандартный раствор, мл |
0 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
|
Поглотительный раствор, мл |
5 |
4,9 |
4,8 |
4,6 |
4,4 |
4,2 |
4,0 |
|
Содержание аммиака, мкг |
0 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
Все пробирки шкалы обрабатывают аналогично пробе. Шкалу стандартов можно использовать для визуального определения, готовят ее одновременно с пробами в колориметрических пробирках.
Расчет количества аммиака см. выше.
Примечания. 1. Все реактивы готовят на дважды перегнанной безаммиачной воде. Приготовление безаммиачной воды см. выше.
2. Так как описанный метод определения аммиака очень чувствителен, посуду необходимо тщательно промывать горячей хромовой смесью, водой, 20% раствором едкого натра и еще раз водой до нейтральной реакции.
ДВУОКИСЬ АЗОТА
|
NO2 |
Мол. вес 46,01 |
|
N2O4 |
Мол. вес 92,02 |
Двуокись азота — желто-бурый газ с резким запахом, легко сгущающийся в жидкость, кипящую при +21 °C, при охлаждении до -11 °C эта жидкость застывает в бесцветную кристаллическую массу. При температуре ниже 140 °C двуокись азота частично полимеризуется в N2O4, и около точки замерзания (-11 °C) вещество состоит только из молекул N2O4. При повышении температуры молекула N2O4 диссоциирует на две молекулы NO2. При растворении NO2, N2O4 в воде образуются азотистая и азотная кислоты.
Двуокись азота раздражающе действует на легкие, вызывая в тяжелых случаях отек их, на верхние дыхательные пути и глаза, а также оказывает общетоксическое действие.
Предельно допустимые концентрации двуокиси азота: максимальная разовая и среднесуточная 0,085 мг/м3.
ОПРЕДЕЛЕНИЕ ДВУОКИСИ АЗОТА [3] С РЕАКТИВОМ ГРИССА — ИЛОСВАЯ
Принцип определения
Метод основан на поглощении двуокиси азота в раствор йодида калия и определения иона нитрита по реакции Грисса — Илосвая.
Чувствительность определения 0,3 мкг двуокиси азота в анализируемом объеме раствора.
Озон не мешает определению до концентраций, превышающих концентрации двуокиси азота в 2 — 3 раза.
Реактивы
1. Поглотительный раствор: 0,5 н. раствор йодида калия.
2. Стандартные растворы нитрита натрия. Исходный стандартный раствор с содержанием 1000 мкг/мл иона ![]() готовят растворением 0,150 г перекристаллизованного нитрита натрия в 100 мл дважды перегнанной воды. Рабочий стандартный раствор с содержанием 1 мкг/мл иона
готовят растворением 0,150 г перекристаллизованного нитрита натрия в 100 мл дважды перегнанной воды. Рабочий стандартный раствор с содержанием 1 мкг/мл иона ![]() готовят разведением исходного раствора в 1000 раз поглотительным раствором.
готовят разведением исходного раствора в 1000 раз поглотительным раствором.
3. Сульфит натрия, 0,01 н. раствор: 0,0630 г безводного сульфита или 0,1261 г Na2SO3·7H2O растворяют в 100 мл дистиллированной воды. Раствор готовят перед анализом.
4. Уксусная кислота (12% раствор).
5. Раствор сульфаниловой кислоты. 0,5 г сульфаниловой кислоты растворяют в 150 мл 12% раствора уксусной кислоты.
6. Раствор ![]() -нафтиламина. 0,2 г
-нафтиламина. 0,2 г ![]() -нафтиламина кипятят с 20 мл воды в течение 2 — 3 мин. до образования на дне капли лилового цвета. Бесцветный раствор декантируют и вливают в 150 мл 12% раствора уксусной кислоты.
-нафтиламина кипятят с 20 мл воды в течение 2 — 3 мин. до образования на дне капли лилового цвета. Бесцветный раствор декантируют и вливают в 150 мл 12% раствора уксусной кислоты.
Растворы сульфаниловой кислоты и ![]() -нафтиламина хранят в темных склянках. Они пригодны к употреблению до тех пор, пока не побуреют.
-нафтиламина хранят в темных склянках. Они пригодны к употреблению до тех пор, пока не побуреют.
7. Реактив Грисса — Илосвая. Перед употреблением смешивают растворы сульфаниловой кислоты и ![]() -нафтиламина в отношении 1:1.
-нафтиламина в отношении 1:1.
Отбор пробы
а) Для определения разовой концентрации воздух со скоростью 0,1 л/мин. протягивают через два поглотительных прибора с пористой пластинкой N 1 с 6 мл поглотительного раствора в каждом.
Продолжительность отбора 20 — 30 мин.
б) Для определения среднесуточной концентрации воздух со скоростью 0,05 л/мин. (3 л/ч) протягивают через два поглотительных прибора, содержащих по 6 мл поглотительного раствора в каждом, отмечая уровень жидкости. Отбор проводят непрерывно в течение суток, периодически добавляя воду до первоначального уровня. Допустимо проводить отбор 6 — 12 раз с перерывами 2 — 4 ч в одни и те же поглотительные приборы.
Ход анализа
В колориметрические пробирки переносят 5 мл пробы из каждого поглотительного прибора, приливают по 0,5 мл реактива Грисса — Илосвая и хорошо перемешивают. Через 10 мин после внесения реактива Грисса — Илосвая непосредственно перед определением добавляют 5 капель 0,01 н. раствора сульфита натрия, взбалтывают и сразу фотометрируют в кюветах с толщиной слоя 1 — 2 см при длине волны 520 нм по сравнению с контролем, который готовят одновременно и аналогично пробам. Содержание двуокиси азота в анализируемом объеме определяют по предварительно построенному калибровочному графику.
Для построения калибровочного графика готовят шкалу стандартов (табл. 20).
Таблица 20
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
|
Рабочий стандартный раствор, мл |
0 |
0,3 |
0,5 |
0,7 |
0,9 |
1,5 |
2 |
|
Поглотительный раствор, мл |
5 |
4,7 |
4,5 |
4,3 |
4,1 |
3,6 |
3 |
|
Содержание двуокиси азота, мкг |
0 |
0,3 |
0,5 |
0,7 |
0,9 |
1,5 |
2 |
Все пробирки шкалы обрабатывают аналогично пробам, измеряют оптическую плотность и строят калибровочный график. Шкалой стандартов можно пользоваться для визуального определения, ее готовят в колориметрических пробирках одновременно с пробами в объеме 2 мл раствора.
Чувствительность определения — 0,1 мкг в анализируемом объеме раствора. В этом случае отбор проб проводят в 2 — 4 мл поглотительного раствора.
Расчет см. выше.
ОПРЕДЕЛЕНИЕ ДВУОКИСИ АЗОТА С ЗАМЕЩЕННЫМ ЭТИЛЕНДИАМИНОМ [2]
Принцип определения
При взаимодействии двуокиси азота в кислой среде с сульфаниловой кислотой образуется диазосоль, которая дает с N-(1-нафтил)-этилендиамином красную окраску, интенсивность которой пропорциональна количеству двуокиси азота.
Чувствительность определения: 0,05 мкг двуокиси азота в 1 мл исследуемого раствора.
Определению мешают сероводород с 0,05 мг/м3 , сернистый газ с 0,4 мг/куб. м, хлор с 0,02 мг/куб. м, формальдегид с 0,08 мг/м3. Закись азота, трехокись азота и нитраты определению не мешают.
Реактивы
1. Поглотительный раствор: 2 мл 0,1% водного раствора HN-(1(нафтил-)-этилендиаминдихлоргидрата доводят в мерной колбе до 100 мл раствором сульфаниловой кислоты. Реактив должен быть бесцветный, его готовят перед анализом.
2. N-(1-нафтил-)-этилендиаминдихлоргидрат, 0,1% водный раствор.
При хранении в темном и прохладном месте устойчив в течение 2 нед.
3. Раствор сульфаниловой кислоты. 5 г сульфаниловой кислоты растворяют в 800 мл дистиллированной воды, подкисляют 140 мл ледяной уксусной кислоты и доводят до 1 л. Раствор следует хранить в темном и прохладном месте, тогда он устойчив в течение 2 нед.
4. Стандартные растворы нитрита натрия.
Исходный стандартный раствор с содержанием 1000 мкг/мл готовят путем растворения 0,3750 г нитрита натрия в 250 мл дистиллированной воды. В темноте и на холоде раствор устойчив в течение 2 нед.
Рабочий стандартный раствор с содержанием 5 мкг/мл. В мерной колбе емкостью 1000 мл разбавляют 5 мл исходного раствора дважды перегнанной водой.
Раствор должен быть свежеприготовленный.
Отбор пробы
а) Для определения разовой концентрации воздух со скоростью 0,2 — 0,3 л/мин. протягивают через два поглотительных прибора со стеклянным фильтром, наполненных по 5 мл поглотительного раствора. Продолжительность отбора 30 мин.
б) Для определения среднесуточной концентрации воздух со скоростью 2,5 л/ч протягивают через два поглотительных прибора, содержащих по 5 мл поглотительного раствора, отмечая уровень жидкости. Отбор проводят непрерывно в течение суток, периодически добавляя воду до первоначального уровня. Допустимо проводить отбор 6 — 12 раз с перерывами 2 — 4 ч в одни и те же поглотительные приборы.
Ход анализа
Содержимое каждого поглотителя доливают дистиллированной водой до прежнего объема и измеряют интенсивность окраски раствора на фотометре в кювете шириной от 1 до 5 см с зеленым светофильтром, с максимумом пропускания 550 нм до сравнению с поглотительным раствором. По калибровочному графику определяют содержание двуокиси азота в 2 мл раствора (табл. 21).
Таблица 21
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
|
Рабочий стандартный раствор, мл |
0 |
0,25 |
0,5 |
1,0 |
1,5 |
2,5 |
|
Поглотительный раствор, мл |
25 |
24,75 |
24,5 |
24,0 |
23,5 |
22,5 |
|
Содержание двуокиси азота, мкг/мл |
0 |
0,05 |
0,1 |
0,2 |
0,3 |
0,5 |
Интенсивность окраски измеряют через 30 мин. с помощью фотометра в кюветах шириной 1 — 5 см с зеленым светофильтром — с максимумом пропускания 550 нм по сравнению с поглотительным раствором. Найденные величины оптической плотности помещают в графике напротив величин содержания двуокиси азота (мкг/мл).
Расчет анализа: <1>
———————————
<1> При расчете анализа надо учитывать, что концентрация двуокиси азота найдена в 1 мл раствора.
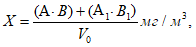
где X — концентрация искомого вещества в миллиграммах на 1 м3 воздуха; A — количество вещества в первом поглотительном приборе в микрограммах; A1 — количество вещества во втором поглотительном приборе в микрограммах; B — объем всей пробы в первом поглотительном приборе в миллиграммах; B1 — объем всей пробы во втором поглотительном приборе в миллиграммах; V0 — объем протянутого воздуха, приведенного к нормальным условиям в литрах.
Примечания. 1. При отборе пробы воздуха поглотительный раствор приобретает красную окраску, пропорциональную количеству двуокиси азота. Второй поглотитель — контрольный. Если наблюдается «проскок» во второй поглотитель, надо уменьшить скорость протягивания воздуха и добавить две капли бутанола.
2. Образующаяся окраска в темноте при комнатной температуре устойчива в течение 48 ч.
3. При использовании кювет с шириной менее 5 см соответственно нужно увеличить концентрацию растворов при построении градуировочной кривой.
РАЗДЕЛЬНОЕ ОПРЕДЕЛЕНИЕ ОКИСИ И ДВУОКИСИ АЗОТА
Принцип определения [1]
Метод основан на окислении окиси азота до двуокиси с помощью окислительной смеси, нанесенной на твердый сорбент, и дальнейшем определении двуокиси азота по образованию азокрасителя с реактивом Грисса — Илосвая.
Чувствительность определения 0,3 мкг в анализируемом объеме раствора.
Озон не мешает определению двуокиси азота до концентраций, превышающих концентрации двуокиси азота в 2 — 3 раза.
Аппаратура
1. V-образная стеклянная трубка, d = 15 — 17 мм, высота — 10 — 12 см.
2. Поглотители с пористой пластинкой N 1.
Реактивы
1. 2,5% раствор серной кислоты.
2. Окислительная смесь — 3% раствор бихромата калия в 2,5% растворе серной кислоты.
3. Твердый сорбент:
а) Инзенский диатомит, размер зерен 0,1 — 0,2 мм, обработанный кипячением в течение 20 — 30 мин. в воде и высушенный на воздухе.
б) Стеклянный порошок, размер зерен 0,25 — 0,5 мм, обработанный кипячением в течение 1 — 1 1/2 ч с соляной кислотой (1:3), промытый водой и высушенный.
4. Твердый сорбент, пропитанный окислительной смесью.
8 — 10 г диатомита или 24 — 25 г стеклянного порошка помещают в фарфоровую чашку, заливают 30 мл окислительной смеси и оставляют на 2 ч. По истечении этого времени раствор сливают, сорбент подсушивают между листами фильтровальной бумаги, сушат в течение 15 — 20 мин. в сушильном шкафу при 50 — 60 °C и досушивают на воздухе. Сорбент, пропитанный окислительной смесью, хранят в склянке с притертой пробкой. Его можно хранить в течение длительного времени.
5. V-образная трубка с твердым сорбентом. V-образную трубку наполняют 6 — 8 г диатомита или 24 — 25 г стеклянного порошка, пропитанного окислительной смесью. В концы трубки вводят тампончики из ваты. Сопротивление трубок должно быть не менее 180 мм вод. ст. Трубка может быть использована на 5 — 6 ч работы.
Приготовление остальных реактивов — см. Определение двуокиси азота выше.
Отбор пробы
Исследуемый воздух со скоростью 0,1 — 0,2 л/мин. протягивают через систему, состоящую из двух поглотительных приборов с пористой пластинкой N 1, содержащих по 5 мл 0,5 н. раствора йодистого калия, и V-образной трубки с твердым сорбентом, пропитанного окислительной смесью (рис. 19). I — поглотительный прибор с 0,5 н. раствором йодистого калия, служащий для поглощения двуокиси азота из воздуха. II — V-образная трубка с твердым сорбентом, пропитанным окислительной смесью, служит для окисления окиси азота до двуокиси. III — поглотительный прибор с 0,5 н. раствором йодистого калия служит для поглощения двуокиси азота, образовавшейся в результате окисления окиси азота.
Рис. 19. Схема присоединения поглотительных
приборов при определении окислов азота
Ход анализа
В колориметрическую пробирку переносят 5 мл пробы, приливают 0,5 мл реактива Грисса — Илосвая и хорошо перемешивают. Через 10 мин. после внесения реактива Грисса — Илосвая непосредственно перед определением добавляют 5 капель 0,01 н. раствора сульфита натрия, взбалтывают и сразу фотометрируют в кюветах с толщиной слоя 1 см при длине волны 520 нм по сравнению с контролем. Содержание двуокиси азота в анализируемом объеме раствора определяют по предварительно построенному градуировочному графику.
Для построения градуировочного графика готовят шкалу стандартов (табл. 22).
Таблица 22
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
|
Рабочий стандартный раствор, мл |
0 |
0,3 |
0,5 |
0,7 |
0,9 |
1,2 |
1,5 |
|
Поглотительный раствор, мл |
5 |
4,7 |
4,5 |
4,3 |
4,1 |
3,8 |
3,5 |
|
Содержание двуокиси азота, мкг |
0 |
0,3 |
0,5 |
0,7 |
0,9 |
1,2 |
1,5 |
Градуировочный график. Шкалу стандартов обрабатывают аналогично пробам и измеряют оптическую плотность. График строят в координатах «концентрация — оптическая плотность». Шкалу стандартов можно использовать для визуального определения.
Расчет анализа
V — общий объем поглотительного раствора в первом поглотительном приборе (мл).
V1 — объем пробы, взятой для анализа из первого поглотительного раствора (мл).
а — количество двуокиси азота в определяемом объеме в микрограммах.
V0 — объем пробы воздуха в литрах (0°, 760 мм рт. ст.).
С1 — концентрация двуокиси азота в первом поглотительном приборе:
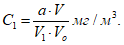
C2 — концентрация двуокиси азота во втором поглотительном приборе рассчитывается по этой же формуле.
Для получения суммы окиси и двуокиси азота (C ) количества двуокиси азота в первом и во втором поглотительных приборах суммируют:
C3 = C1 + C2 мг/м3.
Примечания. 1. При содержании в воздухе больших количеств двуокиси и окиси азота поглощение ведут не в один, а в два поглотительных прибора с йодидом калия.
2. При большой влажности воздуха перед системой ставят трубочку с пемзой, пропитанной серной кислотой.
АЗОТНАЯ КИСЛОТА
Бесцветная жидкость с едким запахом, уд. вес 1,52 (15 °C), температура кипения 86 °C, кипение сопровождается частичным разложением; пары HNO3 в 2,2 раза тяжелее воздуха. Смешивается с водой во всех отношениях, водные растворы полностью диссоциированы. Весьма гигроскопична, сильно дымит на воздухе. HNO3 — сильная кислота, действует на все металлы, кроме золота, платины, иридия и радия. Является сильным окислителем.
Действует раздражающе на дыхательные пути, может вызывать поражения роговицы глаз, разрушение зубов, при попадании на кожу вызывает тяжелые ожоги.
Предельно допустимые концентрации:
а) по молекуле HNO3: максимальная разовая 0,4 мг/м3, среднесуточная 0,4 мг/м3;
б) по водородному иону: максимальная разовая 0,006 мг/м3, среднесуточная 0,006 мг/м3.
ОПРЕДЕЛЕНИЕ АЗОТНОЙ КИСЛОТЫ ПО ИОНУ ВОДОРОДА
Принцип тот же, что и при определении соляной кислоты. Диссоциация азотной кислоты равнозначна соляной кислоте. Определение ее проводят аналогично соляной кислоте (см. выше).
ЛИТЕРАТУРА
1. Дмитриев М.Т., Соловьева Т.В. Раздельное определение окиси и двуокиси азота. Гиг. и сан., 1971, 9.
2. Определение аммиака по реакции с натриевосалицилатным реактивом. Определение аммиака с реактивом Несслера. Определение двуокиси азота с замещенным этилендиамином. В сб.: СЭВ. Унифицированные методы определения атмосферных загрязнений. М., 1970.
3. Полежаев Н.Г., Гирина В.В. Определение двуокиси азота. Гиг. и сан., 1949, 11.
Глава V
СОЕДИНЕНИЯ МЫШЬЯКА, ФОСФОРА, СЕЛЕНА И ТЕЛЛУРА
МЫШЬЯК, МЫШЬЯКОВИСТЫЙ АНГИДРИД И ДРУГИЕ СОЕДИНЕНИЯ МЫШЬЯКА
Мышьяк встречается в нескольких модификациях.
Обычная форма — металлический мышьяк, это кристаллическая масса с металлическим блеском.
Пары мышьяка бесцветны, при резком охлаждении их получается желтый мышьяк, состоящий из прозрачных мягких кубических кристаллов, которые при слабом нагревании, а также под влиянием света переходят в серый мышьяк. Переходным промежуточным продуктом при переходе желтой модификации в металлическую является третья форма мышьяка — черный мышьяк. Мышьяк не растворим в воде.
При нагревании на воздухе мышьяк горит синеватым пламенем, образуя мышьяковистый ангидрид. При горении появляется характерный чесночный запах.
Мышьяковистый ангидрид, называется также белым мышьяком, — белый порошок, растворим в воде, сладковатый раствор его с неприятным металлическим привкусом, имеет слабокислую реакцию. Растворим в щелочах, образует соли.
Соединения мышьяка ядовиты, они поражают нервную систему, органы пищеварения и др.
Предельно допустимая среднесуточная концентрация на мышьяк (неорганические соединения, кроме мышьяковистого водорода в пересчете на мышьяк) 0,003 мг/м3.
ОПРЕДЕЛЕНИЕ СОЕДИНЕНИЙ МЫШЬЯКА
С ДИЭТИЛДИТИОКАРБАМАТОМ СЕРЕБРА [5]
Принцип определения
Соединения мышьяка восстанавливают цинком в солянокислой среде до мышьяковистого водорода, при взаимодействии последнего с раствором диэтилдитиокарбамата серебра в пиридине, раствор окрашивается в красно-фиолетовый цвет.
Чувствительность определения 2 мкг в анализируемом объеме раствора.
Соединения сурьмы мешают определению. Влияние сероводорода устраняют улавливанием его ватой, пропитанной ацетатом свинца.
Аппаратура
1. Прибор для выделения из пробы мышьяковистого водорода. Он состоит из колбы емкостью 50 мл. Колба имеет пришлифованную пробку, в которую впаяны две трубки: верхний конец одной трубки имеет форму воронки, нижний конец трубки доходит до дна. Другая трубка служит для отвода из колбы мышьяковистого водорода.
2. Стеклянная трубка длиной 8 — 9 см наполнена ватой, пропитанной раствором ацетата свинца.
Реактивы
1. Стандартные растворы мышьяка. Исходный раствор с содержанием 100 мкг/мл мышьяка получают растворением в мерной колбе емкостью 100 мл 0,0132 г мышьяковистого ангидрида (сублимированного), 0,1 мл 10% раствора едкого натра и добавляют раствор водой до метки.
Рабочий стандартный раствор с содержанием 2 мкг/мл мышьяка получают разбавлением исходного раствора в 50 раз.
2. Серная кислота, х.ч., не содержащая мышьяка.
3. Азотная кислота, х.ч., 65% раствор.
4. Оксалат аммония, насыщенный раствор.
5. Соляная кислота, 36% раствор, свободная от мышьяка.
6. Йодид калия, х.ч., 1% раствор.
7. Хлорид олова, х.ч., 40% раствор, в соляной кислоте 36% раствор.
8. Цинк зерненный, х.ч., не содержащий мышьяка. Его активируют, обрабатывая разбавленной соляной кислотой.
9. Диэтилдитиокарбамат серебра. 2,25 г диэтилдитиокарбамата натрия растворяют в 100 мл воды и смешивают со 100 мл 1,75% раствора нитрата серебра. Желтый осадок декантируют, отсасывают, промывают водой и высушивают в вакуумном эксикаторе.
Мешает избыток раствора нитрата серебра.
10. Диэтилдитиокарбамат серебра (0,5% раствор в пиридине): 1 г соли растворяют в 200 мл свежеперегнанного пиридина (температура кипения 114 — 116 °C), отфильтрованный раствор сохраняют в темной склянке. Раствор устойчив в течение 2 — 3 мес.
11. Вата, пропитанная ацетатом свинца, для улавливания сероводорода.
В 50 мл воды растворяют 10 г ацетата свинца х.ч., подкисленной уксусной кислотой, прибавляют 20 г глицерина и раствор доводят до 100 мл водой. Вату хлопчатобумажную пропитывают раствором, отжимают избыток и сушат при невысокой температуре. Сохраняют в плотно закрытой банке.
Применяемые реактивы проверяют на отсутствие мышьяка.
Отбор пробы
Для определения среднесуточной концентрации воздух со скоростью 5 л/мин. протягивают непрерывно через фильтр АФА в течение суток. Допустимо проводить отбор 6 — 12 раз с перерывом в 2 — 4 ч в один и тот же фильтр.
Ход анализа
Анализ пробы состоит из трех стадий:
1) минерализация пробы;
2) выделение мышьяковистого водорода;
3) фотометрическое определение мышьяка.
В колбу Кьельдаля емкостью 25 мл помещают фильтр с пробой и по капле смачивают всю поверхность фильтра 1 мл серной кислоты (уд. вес 1,84) и 2 мл 65% раствора азотной кислоты. Колбу медленно нагревают на песчаной бане. Когда фильтр растворится, температуру нагревания повышают. Как только раствор в колбе начнет темнеть, прибавляют азотную кислоту по каплям и продолжают нагревать до выделения белых паров серного ангидрида.
Прозрачный раствор в колбе по охлаждении осторожно разбавляют 3 мл раствора оксалата аммония и раствор снова нагревают до выделения белых паров серного ангидрида. Таким образом устраняют остатки окислов азота, связанных в виде нитрозилсерной кислоты. По охлаждении бесцветный и прозрачный раствор из колбы переводят в мерную колбу емкостью 109 мл или в пробирку с притертой пробкой и меткой — 10 мл. Раствор доводят до метки водой.
Для восстановления соединений мышьяка до мышьяковистого водорода 10 мл раствора из колбы Кьельдаля переводят в реакционную колбу, разбавляют его 10 мл воды, прибавляют постепенно 5 мл соляной кислоты, 2 мл раствора йодида калия и 0,5 мл раствора хлорида олова. Колбу закрывают пробкой и конец изогнутой трубки соединяют с трубкой, наполненной ватой, пропитанной ацетатом свинца. К этой трубке присоединяют поглотительный прибор с пористой пластинкой N 1 с 4 мл пиридинового раствора диэтилдитиокарбамата серебра.
Далее в колбу помещают около 3 г цинка, быстро закрывают, помещают в сосуд с водой (если необходимо подогревание раствора).
Выделение водорода должно быть равномерное: если оно замедляется, то прибавляют через воронку оставшуюся часть соляной кислоты.
Во время восстановительного процесса прибор защищают от солнечных лучей. Процесс восстановления протекает 50 — 60 мин., за это время раствор в поглотительном приборе окрашивается, интенсивность окраски измеряют сразу после окончания восстановления в кювете шириной 2 см по сравнению с контролем, который готовят одновременно и аналогично пробе. Определение проводят при длине волны 533 нм.
Содержание вещества в анализируемом объеме определяют по предварительно построенному калибровочному графику. Для построения калибровочного графика готовят шкалу стандартов (табл. 23).
Таблица 23
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
|
Рабочий стандартный раствор, мл |
0 |
1,0 |
2,0 |
3,0 |
4,0 |
5,0 |
|
Вода, мл |
20 |
19 |
18 |
17 |
16 |
15 |
|
Содержание мышьяка, мкг |
0 |
2 |
4 |
0 |
8 |
10 |
Растворы в реакционных колбах обрабатывают аналогично пробе.
Расчет см. выше.
ОПРЕДЕЛЕНИЕ МЫШЬЯКОВИСТОГО АНГИДРИДА
И ДРУГИХ СОЕДИНЕНИЙ ТРЕХВАЛЕНТНОГО МЫШЬЯКА [3, 8]
Принцип определения
При окислении мышьяковистого ангидрида и других соединений трехвалентного мышьяка смесью перекиси водорода и аммиака образуются соединения пятивалентного мышьяка, которые определяют по синему мышьяково-молибденовому комплексу.
Чувствительность определения 1 мкг в определяемом объеме раствора. Соединения пятивалентного мышьяка, кремния и фосфора мешают определению.
Реактивы
1. Арсенат натрия (трехзамещенный, Na3AsO4 + 2H2O). Исходный стандартный раствор с содержанием 100 мкг мышьяка в 1 мл готовят в мерной колбе емкостью 1 л растворением 0,5661 г соли в воде. Рабочий стандартный раствор с содержанием 1 мкг/мл готовят перед употреблением разбавлением исходного.
2. Молибдат аммония (1% раствор в 5 н. растворе серной кислоты).
3. Гидразин сернокислый (0,15% водный раствор, свежеприготовленный).
4. Аммиак (12% раствор).
5. Перекись водорода (10% раствор).
6. Серная кислота (5 н. раствор).
Отбор пробы
Воздух со скоростью до 50 л/мин. протягивают через фильтр АФА, помещенный в патрон. Отбирают около 1000 л исследуемого воздуха.
Ход анализа
Фильтр помещают в стакан, заливают 8 мл аммиака, добавляют 1 мл раствора перекиси водорода и оставляют на 10 мин., периодически помешивая стеклянной палочкой. Далее раствор переносят в фарфоровую чашку, фильтр промывают раствором аммиака. Смывы объединяют с пробой, выпаривают досуха и сухой остаток растворяют в 2 мл дистиллированной воды. Раствор фильтруют через маленький фильтр в мерную пробирку.
Раствор доводят до объема 5 мл. К пробе добавляют 0,5 мл молибдата аммония, 0,2 мл раствора сернокислого гидразина. Смесь взбалтывают и нагревают на водяной бане в течение 10 — 15 мин.
После охлаждения измеряют оптическую плотность раствора в кювете с толщиной слоя 1 см относительно контроля при длине волны 840 нм. Содержание мышьяка в определяемом объеме устанавливают по предварительно построенному калибровочному графику. Для построения калибровочного графика готовят шкалу стандартов.
На фильтры, употребляемые при отборе проб, наносят различные количества стандартного раствора. Одновременно готовят контроль (табл. 24).
Таблица 24
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
|
Рабочий стандартный раствор, мл |
0 |
1 |
2 |
3 |
4 |
5 |
6 |
|
Содержание мышьяка, мкг |
0 |
1 |
2 |
3 |
4 |
5 |
6 |
Фильтры обрабатывают аналогично пробе, измеряют величины оптических плотностей и строят график.
Шкалой стандартов можно пользоваться также при визуальном определении, ее готовят одновременно с пробой.
Расчет см. выше.
ФОСФОРНЫЙ АНГИДРИД
Кристаллическое или аморфное вещество белого цвета, плотность 2,39, температура плавления 569°, возгоняется при 359 °C. Сильно гигроскопично. Соединяясь с водой, образует фосфорную кислоту. Растворим в серной кислоте.
Фосфорный ангидрид обладает раздражающим и прижигающим действием на слизистые оболочки и кожу.
Предельно допустимые концентрации: максимальная разовая 0,15 мг/м3, среднесуточная 0,05 мг/м3.
ОПРЕДЕЛЕНИЕ ФОСФОРНОГО АНГИДРИДА
С МОЛИБДАТОМ АММОНИЯ [6]
Принцип определения
При взаимодействии фосфорной кислоты с молибдатом аммония образуется фосфорно-молибденовый комплекс, который восстанавливают до фосфорно-молибденовой сини.
Чувствительность определения 0,20 мкг в анализируемом объеме раствора.
Соли мышьяка мешают определению.
Реактивы
1. Стандартный раствор фосфорного ангидрида. Исходный раствор с содержанием 100 мкг/мл фосфорного ангидрида готовят растворением в воде 0,0219 г однозамещенного фосфата натрия или 0,0504 г двузамещенного фосфата натрия в мерной колбе емкостью 100 мл.
Рабочий стандартный раствор с содержанием 1 мкг/мл готовят перед употреблением разбавлением исходного в 100 раз.
Отбор проб, обработку фильтров и приготовление реактивов проводят так же, как при определении мышьяковистого ангидрида по реакции с молибдатом аммония, но без окисления (см. выше).
КАРБОФОС
|
C10H19O6PS2 |
Мол. вес 330,37 |
Маслянистая бесцветная жидкость со слабым неприятным запахом, не растворима в воде, но хорошо растворима в органических растворителях.
Продажный карбофос является 35% концентратом — это густая жидкость от светло-желтого до темно-коричневого цвета с сильным неприятным запахом.
Карбофос — токсичное вещество.
Предельно допустимая концентрация: максимальная разовая — 0,015 мг/м3.
ОПРЕДЕЛЕНИЕ КАРБОФОСА С МОЛИБДАТОМ АММОНИЯ [7]
Принцип определения
При взаимодействии карбофоса со смесью кислот азотной и серной в присутствии перманганата калия происходит разложение карбофоса и выделение фосфорной кислоты, которая с молибдатом аммония образует комплекс. Последний при восстановлении превращается в молибденовую синь, по которой определяют содержание карбофоса.
Чувствительность определения 2,13 мкг карбофоса в анализируемом объеме раствора.
Фосфаты и другие фосфорноорганические соединения мешают определению.
Реактивы
1. Поглотительный раствор: уксусная кислота и вода в отношении 4:1.
2. Стандартный раствор фосфора: исходный раствор с содержанием 100 мкг/мл получают растворением 0,1159 г двузамещенного фосфата натрия подкисленной водой (2 — 3 капли кислоты в мерной колбе емкостью 100 мл).
Рабочие стандартные растворы с содержанием 10 мкг/мл (А) и 1 мкг/мл (Б) получают разбавлением исходного раствора в 10 и 100 раз.
3. Молибдат аммония (0,25% раствор: 0,25 г соли растворяют в 100 мл 1,25 н. раствора серной кислоты).
4. Окислительная смесь: 0,5 г перманганата калия хорошо растирают и растворяют в 10 мл серной кислоты, уд. вес 1,84 (срок годности смеси 5 дней).
5. Восстановительный раствор: 1 г хлорида олова растворяют в 10 мл соляной кислоты, уд. вес 119. Затем 0,1 мл раствора доводят бутанолом до 20 мл. Раствор годен один день.
Отбор пробы
Для определения разовой концентрации воздух со скоростью 0,5 л/мин. протягивают через два поглотительных прибора с пористыми фильтрами N 1, содержащие по 5 мл поглотительного раствора в каждом. Продолжительность отбора 20 — 30 мин.
Ход анализа
Из каждого поглотительного прибора отдельно переносят 4 мл пробы в фарфоровый тигель и вносят 2,5 мл азотной кислоты и 0,1 мл окислительной смеси.
Тигли помещают на слегка подогретую песчаную баню и температуру бани медленно доводят до 80 — 90°. Раствор нагревают до прекращения выделения уксусной кислоты и окислов азота. Затем поднимают температуру бани до 180° (не выше) и раствор выпаривают досуха. По охлаждении остаток растворяют в 1 — 1,5 мл воды. Растворение проводят путем добавления воды по 0,5 мл 2 — 3 раза, переводя раствор в пробирку. Затем наливают 0,3 мл молибдата аммония и через 15 мин. вносят 0,4 мл бутанола. Пробирку плотно закрывают пробкой и содержимое пробирки встряхивают (40 — 50 раз), проводят экстракцию. После разделения слоев отбирают 0,3 мл бутанолового раствора и переносят его в сухую пробирку, в которую вливают 0,9 мл раствора восстановителя.
Через 10 мин. содержание вещества определяют по шкале стандартов, которую готовят одновременно с пробами (табл. 25).
Таблица 25
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
|
Рабочий стандартный раствор, мл (Б) |
0 |
0,2 |
0,3 |
0,4 |
0,5 |
— |
— |
|
Рабочий стандартный раствор, мл (А) |
— |
— |
— |
— |
— |
0,1 |
0,15 |
|
Вода, мл |
1 |
0,8 |
0,7 |
0,6 |
0,5 |
0,9 |
0,85 |
|
Содержание фосфора, мкг |
0 |
0,2 |
0,3 |
0,4 |
0,5 |
1,0 |
1,15 |
|
Содержание карбофоса <1>, мкг |
0 |
2,13 |
3,18 |
4,26 |
5,30 |
10,6 |
12,19 |
———————————
<1> Коэффициент пересчета с фосфора на карбофос 10,67.
Все пробирки шкалы обрабатывают аналогично пробе. Расчет см. выше.
БУТИФОС
Жидкость от желтого до темно-коричневого цвета, температура кипения 154 — 174°. Нерастворима в воде, образует с водой эмульсию, растворима в органических растворителях; обладает сильным неприятным запахом. Раздражает слизистые оболочки носа и глаз.
Предельно допустимые концентрации: максимальная разовая — 0,01 мг/м3, среднесуточная — 0,01 мг/м3.
ОПРЕДЕЛЕНИЕ БУТИФОСА С МОЛИБДАТОМ АММОНИЯ [4]
Метод основан на разложении бутифоса смесью азотной и серной кислот в присутствии перманганата калия и определении выделяющейся фосфорной кислоты с молибдатом аммония.
Чувствительность определения 2 мкг бутифоса в анализируемом объеме раствора.
Реактивы, отбор проб и проведение анализа аналогичны карбофосу (см. выше).
Коэффициент пересчета с фосфора на бутифос 10,1.
Препарат М-81 [7]
(0,0-диметил-![]() -этилмеркаптоэтилдитиофосфат)
-этилмеркаптоэтилдитиофосфат)
|
(CH3O)2PSSCH2CH2SC2H5 |
Мол. вес 246,36 |
Препарат М-81 — бурая жидкость с неприятным запахом, легко смешивающаяся с водой с образованием эмульсии. Применяется как инсектицид, выпускается в виде 50% концентрата в смеси с ОП-7. М-81 обладает высокой токсичностью. Предельно допустимые концентрации: максимальная разовая и среднесуточная 0,001 мг/м3.
Определение препарата М-81 см. «Определение карбофоса» (см. выше).
СЕЛЕН
Селен может быть в аморфном и кристаллическом состоянии. Температура плавления аморфного селена 50°, кристаллического 220°, плотность 4,26 и 4,2 соответственно. Аморфный селен — бурая масса, кристаллический — вещество с металлическим блеском. Селен растворим в воде, в крепком растворе сульфита натрия, в смеси соляной кислоты с бромом, в крепкой серной кислоте. Азотная кислота окисляет селен до селенистого ангидрида. Металлический селен менее ядовит, чем его соединения. Характерные признаки отравления — раздражение слизистых оболочек, общая слабость, головная боль. Предельно допустимая концентрация не установлена.
ОПРЕДЕЛЕНИЕ СЕЛЕНА С 3,31-ДИАМИНОБЕНЗИДИНОМ [1]
Принцип определения
При взаимодействии селена с 3,31-диаминобензидином образуется комплексное соединение, окрашенное в желтый цвет. Чувствительность определения 0,5 мкг в анализируемом объеме раствора. Теллур, медь, железо, мышьяк не мешают определению.
Реактивы
1. Стандартный раствор селена. Исходный раствор селена готовят растворением 500 мг селена при умеренном нагревании на водяной бане в 10 мл соляной кислоты с добавлением 5 — 7 капель азотной кислоты: раствор переводят в мерную колбу емкостью 500 мл и доводят до метки водой. 1 мл раствора содержит 1 мг селена. Рабочий стандартный раствор с содержанием 1 мл/1 мкг селена готовят разбавлением исходного раствора в 1000 раз. Раствор должен быть свежеприготовленным.
2. Соляная кислота, удельный вес 1,19.
3. Азотная кислота, концентрированная.
4. Смесь соляной и азотной кислот, взятых в соотношении 1:1.
5. Комплексон III (динатриевая соль этилендиаминотетрауксусной кислоты) (2,5% раствор).
6. Кислота муравьиная (разбавленная 1:9).
7. Аммиак (разбавленный 1:1).
8. Крезоловый красный (0,1% раствор 100 мг индикатора растворяют в 20 мл 1% раствора едкого натра и доводят до 100 мл).
9. Толуол.
10. 3,31-Диаминобензидин (0,5% раствор). Раствор должен быть свежеприготовленным.
Отбор пробы
Воздух со скоростью 20 л/мин. протягивают через фильтр АФА-В-18, помещенный в патрон. Для анализа следует отобрать 500 л воздуха.
Ход анализа
Фильтр с пробой переносят в стакан и обрабатывают 10 мл смесью соляной и азотной кислот при умеренном нагревании (избегать бурной реакции, которая может привести к улетучиванию селена). Полученный раствор упаривают почти досуха, и нагревание сухого остатка допускать нельзя ввиду возможного улетучивания селена. К осадку добавляют 10 — 15 мл горячей воды и содержимое нагревают до получения прозрачного раствора. К раствору добавляют 2 — 3 капли крезолового красного, 1 мл комплексона III и 2 мл муравьиной кислоты, тщательно перемешивают и нейтрализуют аммиаком до желтой окраски (pH 2,0 — 3,0). К холодному раствору приливают 2 мл свежеприготовленного раствора 3,31-диаминобензидина и ставят в темноту на 30 мин. По истечении времени раствор нейтрализуют аммиаком до появления фиолетовой окраски (pH 8,0), переводят в делительную воронку емкостью 50 — 75 мл и добавляют 5 мл толуола. Воронку энергично встряхивают в течение минуты и после разделения слоев водную фазу отбрасывают, а слой толуола через сухой фильтр сливают в кювету с толщиной слоя 1 см и фотометрируют при длине волны 420 нм по сравнению с контролем. Содержание селена определяют по предварительно построенному калибровочному графику. Для построения калибровочного графика готовят шкалу стандартов, при этом рабочий стандартный раствор наносят на фильтры, которые употребляют при отборе проб. Одновременно готовят контрольный фильтр (табл. 26).
Таблица 26
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
|
Рабочий, стандартный раствор, мл |
0 |
0,5 |
0,7 |
1,0 |
1,2 |
1,5 |
1,7 |
2,0 |
|
Содержание селена, мкг |
0 |
0,5 |
0,7 |
1,0 |
1,2 |
1,5 |
1,7 |
2,0 |
Контрольный фильтр и фильтры, содержащие различное количество селена, обрабатывают аналогично пробам. Шкала стандартов может быть использована для визуального определения. Ее готовят в колориметрических пробирках.
Расчет см. выше.
ТЕЛЛУР
Вещество серебристо-белого цвета. Температура плавления 452 °C, температура кипения 1390 °C. Сгорает с образованием двуокиси теллура. В воде не растворим, не растворим в кислотах, не являющихся окислителями. Растворим в концентрированной серной, азотной кислотах, растворим в царской водке и концентрированных растворах щелочей. Теллур и его соединения токсичны.
Действие их сходно с действием неорганических соединений мышьяка и серы. При попадании теллура в организм через органы дыхания образуется летучее соединение — теллуристый метил, обусловливающий появление специфического чесночного запаха в выдыхаемом воздухе.
ОПРЕДЕЛЕНИЕ ТЕЛЛУРА И ЕГО СОЕДИНЕНИЙ С БУТИЛРОДАМИНОМ С [2]
Принцип определения
При взаимодействии теллура с бутилродамином С образуется окрашенный комплекс, который экстрагируется бензолом.
Чувствительность определения 0,5 мкг теллура в определяемом объеме раствора. Определению мешает медь. Селен определению не мешает.
Реактивы
1. Исходный стандартный раствор с содержанием 1 мг/мл готовят, помещая 0,1 г теллура в выпарительную чашку, в которую добавляют 10 мл концентрированной серной кислоты. Содержимое выпаривают досуха, после охлаждения добавляют 20 мл концентрированной соляной кислоты и переносят в мерную колбу емкостью 100 мл и доводят объем ее до метки водой.
Рабочий стандартный раствор с содержанием 1 мкг/мл готовят соответствующим разбавлением исходного.
2. Серная кислота, уд. вес 1,84, разведенная 1:1.
3. Азотная кислота, уд. вес 1,4.
4. Соляная кислота, уд. вес 1,19; 0,1 и 0,5 н. растворы.
5. Трибутилфосфат, 30% раствор в ксилоле (по объему).
6. Ацетон.
7. Бензол.
8. Бромид натрия (2 М раствор).
9. Бутилродамин С (0,1% водный раствор).
10. Аскорбиновая кислота (2% водный раствор, свежеприготовленный).
Отбор пробы
Воздух со скоростью до 50 л/мин. протягивают через фильтр АФА, помещенный в патрон. Для анализа отбирают не менее 600 — 700 л исследуемого воздуха.
Ход анализа
Фильтр помещают в стакан емкостью 50 мл и заливают смесью концентрированных серной и азотной кислот (1,5:1). После полного растворения фильтра смесь упаривают почти досуха. Влажный остаток обрабатывают 5 мл 5 н. раствором соляной кислоты. Содержимое стакана переносят в делительную воронку емкостью 25 мл. Добавляют 5 мл трибутилфосфата в ксилоле и взбалтывают в течение 2 мин. После разделения к органической фазе добавляют 5 мл 0,1 н. раствора соляной кислоты и реэкстрагируют теллур в течение 2 мин. Полученный реэкстракт осторожно упаривают, не допуская обугливания органики. Остаток обрабатывают 5,5 мл раствора серной кислоты 1:1, добавляют 0,5 мл 2 М раствора бромида натрия, 1 мл 0,1% раствора бутилродамина С и 2 мл раствора аскорбиновой кислоты.
В охлажденную смесь вносят 1 мл ацетона.
Смесь переносят в делительную воронку, в которую вносят 5 мл бензола, и экстрагируют в течение 1 мин. Оптическую плотность бензольного раствора измеряют в кюветах с толщиной слоя 1 см при длине волны 560 нм относительно контроля.
Количество теллура устанавливают по калибровочному графику. Для его построения готовят шкалу стандартов, при этом наносят на фильтры стандартный раствор (табл. 27).
Таблица 27
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
|
Рабочий стандартный раствор, мл |
0 |
0,5 |
1,0 |
1,5 |
2,0 |
2,5 |
|
Содержание теллура, мкг |
0 |
0,5 |
1,0 |
1,5 |
2,0 |
2,5 |
Фильтры с нанесенными стандартными растворами и контроль обрабатывают аналогично пробе. По полученным значениям оптических плотностей строят калибровочный график. Шкалой можно пользоваться при визуальном определении, ее готовят одновременно с пробой.
Расчет см. выше.
ЛИТЕРАТУРА
1. Алексеева Л.С. Спектрофотометрическое определение селена в атмосферном воздухе. Гиг. и сан., 1970, 2.
2. Алексеева Л.С., Мурашова В.И. и др. Фотометрическое определение теллура в атмосферном воздухе. Заводская лаборатория, т. XXXVIII, 11, 1971.
3. Гольдберг Е.Х. Определение мышьяковистого ангидрида и других неорганических соединений мышьяка. Материалы Юбилейной конференции по общей и коммунальной гигиене. М., Санитарно-эпидемиологическая станция г. Москвы, 1967.
4. Мухамедова С.Ю. Экспериментальные данные к нормированию бутифоса в атмосферном воздухе. Гиг. и сан., 1968, 12.
5. Определение соединений мышьяка. В сб. СЭВ: Унифицированные методы определения атмосферных загрязнений. Ч. 1. М., 1970.
6. Определение фосфорного ангидрида. В сб. СЭВ: Унифицированные методы определения атмосферных загрязнений. Ч. II. М., 1973.
7. Технические условия на метод определения карбофоса, метафоса, М-81, октаметила и др. В. III, 1963.
8. Технические условия на метод определения мышьяковистого ангидрида. В. IV, 1965.
Глава VI
МЕТАЛЛЫ
ВАНАДИЙ
Металл серебристо-серого цвета.
Плотность 6,11. Температура плавления 1900 +/- 25 °C.
Температура кипения 3330 °C. Не растворим в воде. Растворим в концентрированных серной, азотной, фтористо-водородной кислотах. Известны окисные соединения: трехокись, пятиокись, ванадат аммония, йодистый ванадий, бромистый ванадий и др.
Соединения ванадия токсичны. Вызывают изменения в органах дыхания, нервной системе, кровообращении, обмене веществ, обладают раздражающим действием.
Данных о вредном действии металлического ванадия не имеется.
Предельно допустимая концентрация пятиокиси ванадия: среднесуточная 0,002 мг/м3.
ОПРЕДЕЛЕНИЕ ВАНАДИЯ С САЛИЦИЛГИДРОКСАМОВОЙ КИСЛОТОЙ [1]
Принцип определения
При окислении соединений ванадия образуется пятиокись ванадия, которая с салицилгидроксамовой кислотой дает ванадий-салицилгидроксамовый комплекс, окрашенный в фиолетовый цвет.
Чувствительность определения — 1 мкг ванадия в анализируемом объеме раствора.
Железо, кальций, магний, марганец, титан, медь, кремний, никель, цинк до 500 мкг не мешают определению.
Реактивы
1. Стандартный раствор ванадия готовят из ванадата аммония NH4VO3. Реактив дважды перекристаллизовывают из горячей воды, охлаждают раствор льдом, отделяют маточный раствор, кристаллы промывают этанолом и сушат на воздухе, затем в сушильном шкафу при 50 °C.
Исходный раствор с содержанием 1 мг/мл готовят растворением 0,2295 г перекристаллизованного ванадата аммония в мерной колбе емкостью 100 мл. Навеску помещают в колбу, растворяют в 50 мл горячей воды (70 — 75°). После растворения и охлаждения раствор доводят до метки.
Разбавляя исходный раствор в 100 раз, получают рабочий стандартный раствор с содержанием 10 мкг/мл.
2. Этиловый спирт.
3. Азотная кислота, уд. вес 1,4, 50% раствор (по объему).
4. Натр едкий, х.ч., 5% раствор; 8,5 и 20% раствор.
5. Уксусная кислота, ледяная.
6. Метиловый эфир салициловой кислоты.
7. Гидроксиламин солянокислый. 35 г гидроксиламина растворяют в 100 мл 20% раствора едкого натра. Хранят в холодильнике или применяют свежеприготовленным.
8. Соляная кислота, уд. вес 1,19.
9. Салицилгидроксамовая кислота, 0,5% раствор в ледяной уксусной кислоте.
Синтез салицилгидроксамовой кислоты: 76 г метилового эфира салициловой кислоты помещают в 300 мл охлажденного 8,5% раствора едкого натра, добавляют 900 мл воды и перемешивают раствор в течение 45 — 50 мин. Затем вносят 100 мл охлажденного раствора гидроксиламина и оставляют на 24 ч. Все операции проводят при температуре не выше 25°. Далее раствор фильтруют, устанавливают величину pH, равную 1 — 2 (по универсальной индикаторной бумаге). Осадок отделяют при помощи воронки Бюхнера, промывают водой, подкисленной соляной кислотой. Выход салицилгидроксамовой кислоты составляет 70 — 72%, температура плавления 140 — 145 °C. Реактив устойчив в течение нескольких лет.
Отбор пробы
А. Для определения разовой концентрации воздух со скоростью 50 — 100 л/мин. протягивают через фильтр АФА, укрепленный в патроне. Продолжительность отбора 20 — 30 мин.
Б. Для определения среднесуточной концентрации воздух со скоростью 10 л/мин. протягивают через фильтр, укрепленный в патроне, непрерывно в течение суток. Допустимо среднесуточный отбор проводить в один и тот же фильтр 6 — 12 раз со скоростью 5 — 10 л/мин. с перерывом в 4 или 2 ч.
Ход анализа
Фильтр извлекают из патрона, обрезают края, переносят в фарфоровый тигель, добавляют 2 мл 50% азотной кислоты, выпаривают досуха на водяной бане или электроплитке, закрытой асбестом. Затем помещают в муфельную печь и постепенно повышают температуру до 500 °C. После полного озоления пробы тигель охлаждают, добавляют 2 мл 5% раствора едкого натра и 5 мл воды. Нагревают тигель в течение 2 — 3 мин. на кипящей водяной бане и содержимое фильтруют в пробирку через маленький фильтр; тигель несколько раз обмывают дистиллированной водой и доводят объем пробы до 10 мл. Берут 5 мл пробы, вносят 5 мл 5% раствора салицилгидроксамовой кислоты, перемешивают. Через 15 — 20 мин. интенсивность окраски измеряют в кювете с толщиной слоя 2 см при длине волны 490 нм по сравнению с контролем. Содержание ванадия во всей пробе определяют по предварительно построенному калибровочному графику.
Для построения калибровочного графика готовят шкалу стандартов, при этом рабочий стандартный раствор наносят на фильтры АФА (табл. 28). Одновременно готовят контрольный фильтр.
Таблица 28
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
|
Рабочий стандартный раствор, мл |
0 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
|
Содержание ванадия, мкг |
0 |
1,0 |
2,0 |
4,0 |
6,0 |
8,0 |
10,0 |
Контрольный фильтр и фильтры, содержащие различные количества ванадия, помещают в тигли и обрабатывают аналогично пробам.
Шкала стандартов может быть использована для визуального определения. Ее готовят в колориметрических пробирках одновременно с пробой.
Коэффициенты пересчета ванадия на:
1. Ванадия окись VO 1,31.
2. Ванадия трехокись V2O3 1,47.
3. Ванадия двуокись VO2 1,62.
4. Ванадия пятиокись V2O5 1,78.
5. Ванадия карбид VC 1,23.
6. Ванадат аммония NH4VO3 2,3.
7. Ванадат калия (мета-) KVO3 2,7. 3
8. Ванадат натрия (мета-) NaVO3 2,39.
Ванадат натрия (орто-) NO2VO4 3,61.
9. Феррованадий FeV 2,23.
Расчет см. выше.
Примечание. При определении только пятиокиси ванадия исключают окисление и фильтры не обрабатывают азотной кислотой.
ХРОМ
Металл серебристо-серого цвета. Плотность 7,16. Температура плавления 1875 °C, температура кипения 2480 °C.
Не растворим в холодной и горячей воде и азотной кислоте, растворим в соляной и серной кислотах.
Соединения хрома могут иметь валентность от двух до шести. В хроматах он трехвалентен, в бихроматах — шестивалентен.
Известны окисные соединения хрома — окись хрома CrO3 зеленого цвета, хромовый ангидрид — желтого цвета, соли трех- и шестивалентного хрома.
Соли шестивалентного хрома легко восстанавливаются в кислой среде до трехвалентного состояния.
Соединения хрома токсичны, особенно ядовиты шестивалентные соединения, меньшей токсичностью характеризуются трехвалентные соединения. Хром и его двухвалентные соединения малотоксичны.
Соединения хрома обладают раздражающим и прижигающим действием, они оказывают также общетоксическое действие. При контакте с хромом отмечена повышенная заболеваемость раком дыхательных путей.
Предельно допустимые концентрации хрома шестивалентного в пересчете на CrO3: максимальная разовая 0,0015 мг/м3, среднесуточная 0,0015 мг/м3.
ОПРЕДЕЛЕНИЕ ХРОМОВОГО АНГИДРИДА И СОЛЕЙ ХРОМОВОЙ
КИСЛОТЫ (Cr6) С ДИФЕНИЛКАРБАЗИДОМ [3]
Принцип определения
При взаимодействии шестивалентного хрома с дифенилкарбазидом в кислой среде происходит образование соединения, окрашенного в красно-фиолетовый цвет.
Чувствительность определения — 0,4 мкг хрома в анализируемом объеме.
Железо, ванадий, молибден мешают определению в количестве более 1 мг.
Реактивы
1. Стандартный раствор хрома. Исходный раствор с содержанием 100 мкг/мл хрома готовят растворением 0,0282 г перекристаллизованного бихромата калия в мерной колбе емкостью 100 мл.
Рабочий стандартный раствор с содержанием 1 мкг/мл готовят перед употреблением разбавлением исходного в 100 раз.
2. Соляная кислота, удельный вес 1,4.
3. Этанол.
4. Смесь 1 мл соляной кислоты и 100 мл этанола.
5. Дифенилкарбазид. 0,5 г дифенилкарбазида растворяют во всем объеме смеси соляной кислоты с эталоном.
Отбор пробы
А. Для определения разовой концентрации воздух со скоростью до 50 л/мин. протягивают через фильтр АФА, беззольный фильтр, укрепленный в патроне. Продолжительность отбора 20 — 30 мин.
Б. Для определения среднесуточной концентрации воздух со скоростью 10 — 15 мин. протягивают через фильтр, укрепленный в патроне, в течение суток непрерывно. Допустимо отбор проб проводить 6 — 12 раз в один и тот же фильтр с перерывами в 2 или 4 ч.
Ход анализа
Фильтр извлекают из патрона, обрезают края, помещают в стакан и заливают 5 мл горячей воды. Помешивают стеклянной палочкой. Через 10 — 15 мин. 4 мл прозрачного раствора переносят в пробирку, вносят 0,4 мл раствора дифенилкарбазида, перемешивают. Через 10 — 15 мин. интенсивность окраски измеряют при помощи фотометра в кювете с толщиной слоя в 1 или 2 см при длине волны 540 нм по сравнению с контролем. Содержание хрома в анализируемом объеме (4 мл) определяют по предварительно построенному калибровочному графику. Для построения калибровочного графика готовят шкалу стандартов. При этом рабочий стандартный раствор наносят на фильтры, употребляемые для отбора проб (табл. 29). Одновременно готовят и контрольный фильтр.
Таблица 29
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
|
Рабочий стандартный раствор, мл |
0 |
0,4 |
0,6 |
0,8 |
1,0 |
1,2 |
1,5 |
|
Содержание хрома, мкг |
0 |
0,4 |
0,6 |
0,8 |
1,0 |
1,2 |
1,5 |
Коэффициент пересчета с хрома на CrO3 1,94.
Контрольный фильтр и фильтры, содержащие различные количества хрома, помещают в стаканы, обрабатывают аналогично пробе.
Шкала стандартов может быть использована для визуального определения. Ее готовят в колориметрических пробирках одновременно с пробами, как указано в табл. 29.
Расчет см. выше.
ОПРЕДЕЛЕНИЕ СОЕДИНЕНИЙ ДВУХ- И ТРЕХВАЛЕНТНОГО ХРОМА [5]
Принцип определения
Соединения двух- и трехвалентного хрома окисляют перманганатом калия. Образующиеся соединения шестивалентного хрома при взаимодействии с дифенилкарбазидом дают окрашенные в розово-фиолетовый цвет соединения.
Чувствительность определения — 1 мкг шестивалентного хрома в анализируемом объеме раствора. Определению мешают железо, ванадий, медь, молибден, ртуть в количествах, во много раз превышающих содержание хрома.
Реактивы
1. Азотная кислота (1,4), разбавленная 1:1, 2 н. раствор.
2. Серная кислота (1,84), 0,5 н. раствор.
3. Перманганат калия, 0,2 н. раствор.
4. Фосфорная кислота, концентрированная.
5. Этиловый спирт, 96°.
6. Аммиак, 25% раствор.
7. Дифенилкарбазид, 0,5% раствор. Готовят в мерной колбе емкостью 100 мл, в которую вносят 0,5 г дифенилкарбазида, 1 мл концентрированной соляной кислоты, доводят до метки этанолом.
Приготовление стандартного раствора и отбор пробы см. «Определение шестивалентного хрома».
Ход анализа
Фильтр заливают 40 мл 2 н. раствором азотной кислоты и нагревают на песчаной бане в течение 15 мин. Раствор фильтруют, промывают фильтр 2 н. раствором азотной кислоты и водой. Фильтрат выпаривают и пробу помещают в муфельную печь. Температура печи не должна превышать 550°. Сжигание проводят в течение 30 мин. Далее пробу растворяют в 7 мл 0,5 н. раствора серной кислоты и подогревают на водяной бане. Нерастворимый остаток отфильтровывают. Ставят пробу на водяную баню, нагретую до 100 °C, и добавляют по каплям 0,2 н. раствора перманганата калия до получения устойчивого розового окрашивания. Нагревают в течение 15 мин. и вводят по каплям этиловый спирт до обесцвечивания. Пробу переносят в мерную колбу емкостью 25 мл, добавляют 1 мл фосфорной кислоты и дистиллированной воды до объема около 20 мл. Затем добавляют 1 мл раствора дифенилкарбазида и доводят объем колбы до метки. Измеряют оптическую плотность в кюветах с шириной слоя 1 см при длине волны 540 нм. Калибровочный график строят на основании приготовленной шкалы стандартов. Для этого на фильтры, употребляемые при отборе, наносят стандартный раствор. Фильтры обрабатывают аналогично пробе, одновременно готовят контрольный фильтр.
Шкалу строят с содержанием от 1 до 30 мкг хрома.
Примечание. Влияние железа устраняют добавлением фосфорной кислоты. Влияние остальных металлов можно устранить экстрагированием хлороформом, за исключением ртути, которую связывают добавлением раствора хлористого натрия. В случае присутствия значительных количеств железа (более 1000 мкг) его осаждают. Для этой цели после добавления этанола для восстановления перманганата калия в пробу вводят раствор аммиака до щелочной реакции. При подогревании образуется гидроокись железа, которую удаляют фильтрованием. Аммиак удаляют подогреванием до получения нейтральной реакции. Далее анализ проводят, как указано выше.
Расчет см. выше.
МАРГАНЕЦ
Металл серовато-серебристого цвета. Окисная пленка сообщает металлу розовую окраску. Плотность 7,2.
Температура плавления 1244 °C, температура кипения 2120 °C.
Растворим в уксусной и минеральных кислотах. Соединения марганца могут иметь валентность от 2+ до 7+.
Известны окисные соединения марганца. Закись — MnO, окись — Mn3O4 , двуокись — MnO2, марганцовистый ангидрид — Mn2O7. Водные растворы марганцовистой кислоты окрашены в зеленый цвет, марганцовой — в фиолетовый.
Соединения марганца токсичны, действуют на центральную нервную систему, легкие, печень, вызывают изменения состава крови.
Предельно допустимая концентрация марганца и его соединений в пересчете на MnO2: среднесуточная 0,01 мг/м3.
ОПРЕДЕЛЕНИЕ МАРГАНЦА С ПЕРСУЛЬФАТОМ АММОНИЯ [8]
Принцип определения
При взаимодействии иона марганца с персульфатом аммония в присутствии нитрата серебра, играющего роль катализатора, происходит окисление марганца с образованием марганцовой кислоты, окрашенной в фиолетовый цвет.
Чувствительность определения — 2 мкг в определяемом объеме раствора.
Хром мешает определению.
Реактивы
1. Стандартный раствор марганца: исходный раствор с содержанием 100 мкг/мл марганца готовят растворением 0,0504 г сульфата марганца MnSO4·7H2O в 5% растворе серной кислоты в мерной колбе емкостью 100 мл.
Рабочий стандартный раствор с содержанием 1 мкг/мл готовят разбавлением исходного в 100 раз.
2. Серная кислота, уд. вес 1,82 — 1,84, 5% раствор.
3. Щавелевая кислота, 8% раствор.
4. Смесь кислот серной (уд. вес 1,82 — 1,84) и 8% щавелевой в отношении 1:1 готовят перед употреблением.
5. Персульфат аммония, кристаллический.
6. Нитрат серебра, 0,1% раствор.
7. Ортофосфорная кислота, 20% раствор.
Отбор пробы
А. Для определения разовой концентрации воздух со скоростью до 50 л/мин. протягивают через фильтр АФА, беззольный, укрепленный в патроне. Продолжительность отбора 20 — 30 мин.
Б. Для определения среднесуточной концентрации исследуемый воздух со скоростью до 10 л/мин. протягивают через фильтр, укрепленный в патроне, в течение суток непрерывно.
Допустимо среднесуточный отбор проводить в один и тот же фильтр 6 — 12 раз с перерывами в 4 или в 2 ч.
Ход анализа
Фильтр извлекают из патрона, обрезают края, помещают в фарфоровую чашку. Осторожно озоляют при 500 — 600 °C. К озоленной пробе добавляют 0,5 мл смеси кислот, помещают чашку на песчаную баню и нагревают до удаления пузырьков углекислоты и белых паров серного ангидрида.
Образовавшийся сульфат марганца растворяют в 1 мл 5% раствора серной кислоты. Раствор фильтруют через воронку диаметром 2 — 3 см с оттянутым концом, в который вложен тампон, состоящий из волокон бумажного фильтра, уложенных длиной около 0,5 см. Чашку тщательно обмывают несколькими порциями 5% раствора серной кислоты, растворы фильтруют.
Общий объем пробы доводят до 4 мл. Для анализа берут до 3,5 мл пробы. Можно в чашку сразу добавить 4 мл 5% серной кислоты, перенести раствор в центрифужные пробирки и центрифугировать, и в этом случае для анализа берут до 3,5 мл прозрачного раствора. Вносят 0,1 мл нитрата серебра, по 0,1 мл ортофосфорной кислоты и добавляют несколько кристалликов персульфата аммония; перемешивают и пробирки с пробами помещают на водяную баню, нагретую до 70 °C на 5 мин. После охлаждения интенсивность окраски измеряют при помощи фотометра в кювете с толщиной слоя 1 или 2 см при длине волны 545 пм по сравнению с контролем. По предварительно построенному калибровочному графику устанавливают содержание марганца в анализируемом объеме (4 мл).
Для построения калибровочного графика готовят шкалу стандартов. При этом рабочий стандартный раствор наносят на такие же фильтры, как и употребляемые для отбора пробы (табл. 30). Одновременно обрабатывают и контрольный фильтр.
Таблица 30
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
|
Рабочий стандартный раствор, мл |
0 |
2 |
3 |
5 |
7 |
9 |
|
Содержание марганца, мкг |
0 |
2 |
3 |
5 |
7 |
9 |
Контрольный фильтр и фильтры, содержащие различные количества марганца, помещают в фарфоровые чашки, озоляют. Контрольный фильтр и шкалу обрабатывают аналогично пробам. Шкала стандартов может быть использована для визуального определения. Ее готовят в колориметрических пробирках, одновременно с пробами, так же, как указано в табл. 30.
Коэффициент пересчета с Mn на MnO2 1,58.
Расчет см. выше.
КОБАЛЬТ
Металл серебристо-серого цвета. Плотность 8,8. Температура плавления 1492 °C, температура кипения 2255 °C. Растворим в азотной, соляной кислотах. Серная кислота не действует на кобальт на холоде. Стоек по отношению к воздуху и воде. Кобальт и его соединения токсичны. Действуют на верхние дыхательные пути, почки, вызывают тромбоз сосудов и др.
НИКЕЛЬ
Металл серебристо-белого цвета. Плотность 8,9. Температура кипения 2140 °C, температура плавления 1453 °C. Плохо растворим в воде, растворим в разбавленной азотной, соляной, серной кислотах. Никель относится к мало активным элементам, окисляется лишь при 500 °C. Никель и его соединения токсичны. Аэрозоли их раздражают верхние дыхательные пути, изменяют состав крови.
МЕДЬ
Металл бледно-розового цвета. Плотность 8,92. Температура плавления 1083 °C, температура кипения 2580 °C. Растворима в азотной и концентрированной серной кислотах. Реагирует с соляной кислотой и гидратом окиси аммония. На воздухе окисляется, образуя окисную пленку. Соединения меди токсичны. Действуют на кожные покровы, слизистые оболочки, вызывая высыпания, зуд, головную боль, лихорадку.
ОПРЕДЕЛЕНИЕ МЕДИ, КОБАЛЬТА И НИКЕЛЯ ХРОМАТОГРАФИЕЙ
НА БУМАГЕ [4]
Принцип определения
Медь, кобальт, никель переводятся в растворимые соединения обработкой крепкими кислотами и разделяются на бумаге в системе бутанол — ацетон — соляная кислота — вода.
Для проявления разделенных зон применяют рубеано-водородную кислоту и аммиак. Медь окрашивается в серо-зеленый, кобальт — в желтый, никель — сине-фиолетовый цвета.
Чувствительность определения — 0,1 мкг кобальта и никеля и 0,2 мкг меди на хроматограмме.
Определению не мешают алюминий, цинк, олово, марганец.
Влияние железа устраняется в процессе обработки пробы, свинец маскируется в процессе проявления хроматограммы.
Аппаратура
1. Спектрофотометр СФ-10, СФ-14. Плосковыпуклые линзы с оптической силой +10D.
2. Хроматографическая камера (эксикатор).
Реактивы
1. Соляная кислота концентрированная, х.ч., 7 н. раствор, 2 н. раствор, 0,1 н. раствор, 7 н. раствор, насыщенный диэтиловым эфиром. Последний раствор готовят следующим образом. 7 н. раствор соляной кислоты охлаждают до 10 °C, встряхивают с небольшой порцией эфира и снова охлаждают. Эту операцию повторяют до тех пор, пока над кислотой не останется слой эфира. Смесь готовят перед употреблением.
2. Азотная кислота, концентрированная, х.ч.
3. Эфир диэтиловый (для наркоза).
4. Спирт бутиловый, нормальный, х.ч.
5. Ацетон, х.ч.
6. Спирт этиловый, перегнанный при 78 °C.
7. Серная кислота, концентрированная, х.ч.
8. Рубеановодородная кислота, 0,1% раствор в этиловом спирте.
9. Виннокаменная кислота, 4% раствор в этиловом спирте.
10. Аммиак, 25% раствор.
11. Бумага хроматографическая «средняя».
12. Система растворителей. Спирт бутиловый 76 мл, ацетон 36 мл, соляная кислота концентрированная 44 мл и 14 мл воды вносят в делительную воронку и перемешивают.
13. Стандартный раствор никеля. Исходный раствор готовят растворением 0,4049 г хлорида никеля в 100 мл 7 н. раствора соляной кислоты. 1 мл раствора содержит 1 мг никеля. Рабочий стандартный раствор готовят соответствующим разбавлением 7 н. раствором соляной кислоты. Рабочий стандартный раствор содержит 100 мкг/мл.
14. Стандартный раствор кобальта. Исходный раствор с содержанием кобальта 1 мг/мл готовят растворением 0,4037 г хлорида кобальта в 100 мл 7 н. раствора соляной кислоты. Рабочий стандартный раствор с содержанием 100 мкг кобальта в 1 мл готовят соответствующим разбавлением исходного раствором 7 н. соляной кислоты.
15. Стандартный раствор меди. Исходный раствор с содержанием меди 1 мг/мл готовят растворением 0,2667 г хлорида меди в 100 мл 7 н. раствора соляной кислоты. Рабочий стандартный раствор с содержанием 100 мкг меди в 1 мл готовят соответствующим разбавлением 7 н. раствором соляной кислоты.
16. Смесь стандартных растворов с содержанием по 10 мкг меди, никеля и кобальта в объеме 1 мл. В мерную колбу емкостью 25 мл вносят по 2,5 мл рабочих стандартных растворов меди, кобальта, никеля. Объем колбы доводят до метки 7 н. раствором соляной кислоты.
Отбор пробы
Исследуемый воздух со скоростью 50 л/мин. протягивают через фильтр АФА, укрепленный в патроне, в течение 20 — 30 мин.
Ход анализа
Фильтр переносят в фарфоровый тигель, добавляют 1,5 мл концентрированной соляной кислоты, 0,5 мл азотной кислоты (1,4) и нагревают в течение 5 — 10 мин., не допуская кипения. Затем добавляют 0,25 мл серной кислоты и выпаривают досуха. Сухой остаток помещают в муфельную печь, постепенно доводят температуру до 500°.
После охлаждения зольный остаток растворяют в 1,5 мл соляной кислоты (1,19), вносят 0,5 мл азотной кислоты (1,4 уд. вес) и выпаривают досуха при слабом нагревании.
Остаток обрабатывают 3 мл 7 н. раствора соляной кислоты и переносят в делительную воронку. Тигель обмывают 7 н. раствором соляной кислоты, доводят объем до 5 мл. Затем в делительную воронку вносят 5 мл диэтилового эфира, встряхивают в течение 2 — 3 мин. После разделения слоев нижний сливают в тигель. Верхний слой, эфирный, промывают 1 мл 7 н. раствором соляной кислоты, насыщенным эфиром. Полученный после промывания водный слой сливают в тигель.
Если эфирный слой окрашен в желтый цвет, то водный слой подвергают повторной обработке до тех пор, пока экстрагент не станет бесцветным. Далее водный слой, находящийся в тигле, выпаривают до получения влажного остатка, который обрабатывают 0,5 мл 0,1 н. раствора соляной кислоты.
На листе хроматографической бумаги шириной 95 мм и длиной 220 мм на расстоянии 18 мм от нижнего края во всю ширину намечается линия старта. От нее вверх вырезают полосы шириной 6,5 мм и высотой 80 мм. Расстояние между полосами 10 мм. Для очистки бумаги ее помещают в хроматографическую камеру, на дно которой вносят 2 н. раствор соляной кислоты. После поднятия кислоты на высоту 150 мм бумагу вынимают и высушивают. Хранят в бумажном пакете не более 6 дней.
На бумагу наносят 0,05 мл пробы, обдувая ее воздухом, подогретым теплоэлектровентилятором. Разделение металлов проводят в хроматографической камере, на дно которой за 10 мин. до хроматографирования вносят систему растворителей. Бумагу закладывают так, чтобы нижний край погружался в растворитель на 1 см. Когда растворитель продвинется на высоту 8 см от линии старта, бумагу извлекают, высушивают на воздухе и проявляют раствором виннокаменной кислоты путем двустороннего орошения, а затем через 1 — 2 мин. раствором рубеановодородной кислоты с последующей обработкой парами аммиака в течение 2 — 3 мин. При наличии меди наблюдается серо-зеленая зона Rf 0,75, зона кобальта окрашена в желтый цвет Rf 0,40, сине-фиолетовая зона характерна для никеля Rf 0,25.
Количественное определение меди, кобальта, никеля проводят непосредственно на хроматограмме с помощью спектрофотометра СФ-10 или СФ-14. Для этого на входе лучей в интегрирующий шар справа и слева по ходу лучей ставят плосковыпуклые линзы с оптической силой +10D, концентрируя световой пучок до размеров 6,5 на 15 мм. Проводят измерение величины отражения со всей площади окрашенной зоны. В качестве фона для сравнения берут две контрольные полоски бумаги и помещают в кюветы, расположенные справа и слева в нижней части интегрирующего шара. Записывают отражение фона. Затем вместо правого по ходу лучей эталона помещают измеряемый образец и записывают спектр отражения, по которому далее находят область минимального значения отражения каждого элемента. При этой длине волны вычисляют коэффициенты отражения (Rотр.) для меди, кобальта, никеля по формуле:
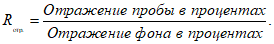
Для построения калибровочного графика готовят шкалу стандартов. При этом смесь стандартных растворов меди, кобальта и никеля наносят на фильтры, употребляемые для отбора пробы (табл. 31).
Таблица 31
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
|
Смесь стандартных растворов никеля, кобальта, меди по 10 мкг, мл |
0 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
|
Соляная кислота 7 н., мл |
1 |
0,9 |
0,8 |
0,6 |
0,4 |
0,2 |
0 |
|
Содержание никеля, кобальта, меди, мкг |
0 |
1 |
2 |
4 |
6 |
8 |
10 |
Фильтры с нанесенными стандартными растворами помещают в тигли, добавляют 0,25 мл серной кислоты и содержимое выпаривают досуха. Пробы озоляют при 500 °C. Далее шкалу стандартов обрабатывают аналогично пробе.
Калибровочный график строят с применением метода Кортюма в координатах:
на оси ординат 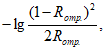
на оси абсцисс ![]()
Количественное определение меди, кобальта, никеля может быть также проведено методом стандартных серий путем сравнения интенсивности зон локализаций пробы со шкалой стандартов.
Пример расчета:
Анализируемая проба рубеаната никеля при длине волны 610 нм имеет величину отражения 65%, отражение фона составляет 100%.
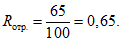
Вычисляется значение функции:
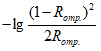
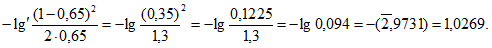
По калибровочному графику величина 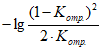 на оси ординат, равная 1,0269, соответствует на оси абсцисс величине -lgC = 0,18.
на оси ординат, равная 1,0269, соответствует на оси абсцисс величине -lgC = 0,18.
lgC = -0,18 = 1,82. По таблицам антилогарифмов находят значение концентрации С = 0,66 мкг.
ЦИНК
Металл серебристого цвета.
Плотность 7,14.
Температура плавления 419,505 °C, температура кипения 913 °C.
Не растворим в воде. Хорошо растворяется в слабых минеральных кислотах, концентрированных щелочах. Легко окисляется на воздухе, образуя окись цинка.
Соединения цинка токсичны. Растворимые соли цинка обладают прижигающим действием.
Окись цинка вызывает катаральное заболевание дыхательных путей и пищеварительных органов. Вдыхание паров окиси цинка связано с литейной лихорадкой.
ОПРЕДЕЛЕНИЕ ЦИНКА С СУЛЬФАРСАЗЕНОМ [2]
Принцип определения
При взаимодействии иона цинка с сульфарсазеном в среде винной, лимонной кислот и аммиака (pH 9,6) образуется комплексное соединение, окрашенное в оранжево-красный цвет.
Чувствительность — 0,5 мкг в определяемом объеме раствора.
Медь, свинец, никель, кобальт, марганец удаляют в процессе анализа, так как они мешают определению.
Железо3, ванадий4, молибден, алюминий, литий не мешают определению в количествах до 1 мг.
Реактивы
1. Исходный стандартный раствор с содержанием 1 мг цинка в 1 мл готовят растворением 0,025 г металлического (химически чистого) цинка в мерной колбе емкостью 25 мл; объем доводят до метки раствором соляной кислоты (1:1).
Стандартный раствор А с содержанием 0,1 мг цинка в 1 мл готовят следующим образом: 2,5 мл исходного стандартного раствора вносят в мерную колбу емкостью 25 мл, нейтрализуют 5% раствором едкого натра по лакмусу. Объем доводят до метки водой.
Рабочий стандартный раствор с содержанием 0,01 мг цинка в 1 мл готовят разбавлением водой раствора А в 10 раз.
2. Едкий натр, 5% раствор.
3. Соляная кислота, уд. вес 1,19 (1:1).
4. Диэтилдитиокарбамат натрия, 1% раствор.
5. Серная кислота, 0,1 н. раствор.
6. Аммиак, 5% раствор. 5 мл концентрированного аммиака помещают в мерную колбу емкостью 100 мл, доводят до метки водой. 10 мл раствора титруют 0,1 н. раствора серной кислоты в присутствии метилового оранжевого, до перехода желтой окраски в розовую. 1 мл 0,1 н. раствора серной кислоты соответствует 0,001703 г аммиака. Соответствующим разбавлением готовят 5% раствор аммиака.
7. Винная кислота, 10% раствор.
8. Лимонная кислота, 10% раствор.
9. Сульфарсазен, 0,02% водный раствор (свежеприготовленный).
10. Сульфат меди. Раствор с содержанием 1 мг меди в 1 мл готовят растворением 0,393 г сульфата меди в мерной колбе емкостью 100 мл, объем до метки доводят водой.
Отбор пробы
А. Для определения разовой концентрации воздух со скоростью 10 — 15 л/мин. протягивают через фильтр АФА или бумажный фильтр, укрепленный в патроне. Продолжительность отбора 20 — 30 мин.
Б. Для определения среднесуточной концентрации воздух со скоростью 2 — 5 л/мин. протягивают через фильтр, укрепленный в патроне, непрерывно в течение суток.
Допустимо среднесуточный отбор проводить в один и тот же фильтр 6 — 12 раз с перерывом в 4 или 2 ч.
Ход анализа
Фильтр извлекают из патрона, обрезают края, помещают в фарфоровую чашку, в которую вносят 10 мл раствора соляной кислоты 1:1, и кипятят в течение 5 мин.
Фильтр отжимают, промывают водой и удаляют. Промывные жидкости объединяют в фарфоровой чашке. В чашку вносят 200 мкг сульфата меди для соосаждения сопутствующих металлов (меди, свинца, кобальта, марганца, никеля), содержимое чашки упаривают досуха на водяной бане. Сухой остаток растворяют в 1,6 мл 5% раствора аммиака и добавляют 0,3 мл 1% раствора диэтилдитиокарбамата натрия. Перемешивают и добавляют 1,3 мл воды. Выделившийся осадок диэтилдитиокарбаматов металлов фильтруют через воронку диаметром 2 — 3 см с оттянутым концом, в которую вложен тампон, состоящий из волокон бумажного фильтра, уложенных длиной около 0,5 см.
Для анализа берут 1,6 мл пробы, добавляют 1,1 мл воды, 0,8 мл 10% раствора винной кислоты, 0,2 мл 10% раствора лимонной кислоты, 0,3 мл 5% раствора аммиака и 1 мл 0,02% раствора сульфарсазена. Раствор перемешивают и интенсивность окраски измеряют при помощи фотометра в кювете с толщиной слоя 1 см при длине волны 500 нм по сравнению с контролем.
По предварительно построенному калибровочному графику устанавливают содержание цинка в анализируемом объеме пробы.
Для построения калибровочного графика готовят шкалу стандартов, при этом рабочий стандартный раствор наносят на фильтры, которыми пользуются при отборе проб. Одновременно готовят контрольный фильтр (табл. 32).
Таблица 32
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
|
Рабочий стандартный раствор, мл |
0 |
0,05 |
0,1 |
0,3 |
0,5 |
0,7 |
1,0 |
|
Содержание цинка, мкг |
0 |
0,5 |
1,0 |
3,0 |
5,0 |
7,0 |
10 |
Контрольный фильтр и фильтры, содержащие различное количество цинка, обрабатывают аналогично пробам. Шкала стандартов может быть использована для визуального определения. Ее готовят в колориметрических пробирках, как указано в табл. 32.
Расчет см. выше.
РТУТЬ
Серебристый жидкий металл.
Плотность 13,546. Температура плавления 38,87 °C, температура кипения 356,58 °C.
В воздухе может находиться в виде паров, причем концентрация ртути меняется с переменой давления и температуры.
Максимально возможная концентрация ртути: при 20 °C и давлении 0,0013 мм 15,2 мг/м3, при 30 °C и давлении 0,0029 мм 33,9 мг/куб. м, при 40 °C и давлении 0,0060 мм 70 мг/м3.
Пары ртути в 7 раз тяжелее воздуха.
Ртуть очень мало растворима в воде, растворяется в растворах хлористого натрия, азотной, серной кислотах, царской водке. Образует амальгамы с рядом металлов, оловом, цинком, свинцом и др.
Ряд соединений ртути под действием восстановителей легко разлагается с выделением металлической ртути.
Пары ртути и ее соединения ядовиты, вызывают нарушения функции нервной системы, почек, печени, обладают кумулятивным действием.
Предельно допустимая концентрация ртути металлической: среднесуточная 0,0003 мг/м3.
ОПРЕДЕЛЕНИЕ РТУТИ [6]
Принцип определения
При взаимодействии ртути с йодом образуется йодистая ртуть, которая с двуххлористой медью и сульфатом натрия образует взвесь, представляющую собой комплексное соединение  окрашенное в красный цвет. Одновременно образуется белая взвесь CuI, служащая фоном, благодаря чему осадок в присутствии ртути окрашен от желтовато-розового до красновато-оранжевого цвета.
окрашенное в красный цвет. Одновременно образуется белая взвесь CuI, служащая фоном, благодаря чему осадок в присутствии ртути окрашен от желтовато-розового до красновато-оранжевого цвета.
Чувствительность определения 0,02 мкг в определяемом объеме раствора. Определению мешает железо.
Реактивы
1. Йод кристаллический.
2. Йодид калия, х.ч.
3. Поглотительный раствор. Готовят из 3 г йодида калия, в который добавляют 3 — 5 мл воды и вносят 0,25 г йода. После растворения йода доводят объем дистиллированной водой до 1 л.
4. Исходный стандартный раствор с содержанием 100 мкг ртути в 1 мл готовят растворением 0,0135 г хлорида ртути или 0,0226 г йодида ртути в мерной колбе емкостью 100 мл. Объем до метки доводят поглотительным раствором.
Рабочий стандартный раствор с содержанием 0,1 мкг в 1 мл готовят разбавлением исходного в 1000 раз поглотительным раствором.
5. Хлорид меди, 7% раствор.
6. Сульфит натрия, х.ч., 3 н. раствор, 0,1 н. раствор, 37,8 г сульфита натрия растворяют в 100 мл воды. Титр 3 н. раствора сульфита устанавливают по 0,1 н. раствору йода. Раствор должен быть свежеприготовленный, так как сульфит натрия легко окисляется на воздухе в сульфат натрия. 0,1 н. раствор готовят разбавлением 3 н. раствора в 30 раз.
7. Смесь хлорида меди и сульфита натрия. Один объем 7% раствора хлорида меди вносят в мерный цилиндр, добавляют 5 объемов сульфита натрия. Осторожно перемешивают стеклянной палочкой до полного растворения осадка. Раствор должен быть прозрачным.
Отбор пробы
А. Для определения разовой концентрации воздух протягивают со скоростью 1 л/мин. через два последовательно соединенных поглотительных прибора Полежаева, наполненных по 1 мл поглотительного раствора.
Перед отбором проб к входному отверстию поглотительного прибора присоединяют компенсатор йода.
Компенсатор представляет собой сосуд, через который проходит впаянная стеклянная трубка с отверстием. Сбоку впаяна вторая трубка, через которую в сосуд перед отбором проб вносят 0,05 г кристаллического йода и отверстие закрывают. Отверстие в стеклянной трубке, проходящей внутри сосуда, служит для поступления йода в поглотительный раствор для уравнивания его концентрации.
Компенсатор присоединяют к системе поглотителей при помощи отрезка каучуковой трубки так, чтобы отверстие во внутренней трубке было обращено вверх (рис. 20).
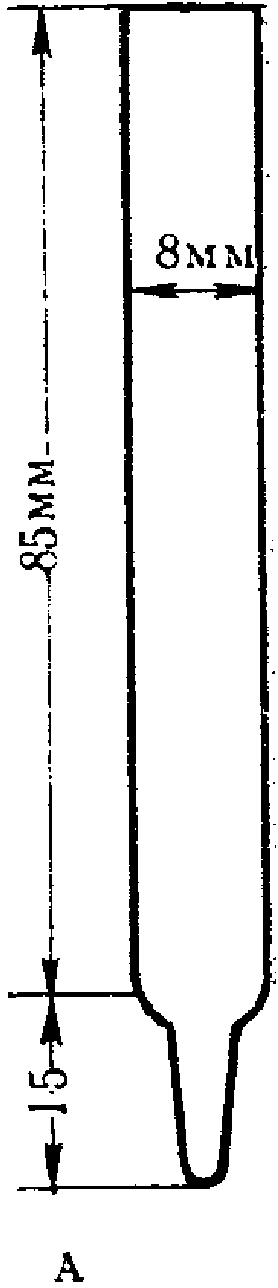
Рис. 20. Схема присоединения компенсатора йода
к поглотительным приборам. А — пробирка
для определения ртути
Продолжительность отбора пробы один час.
Б. Для определения среднесуточной концентрации воздух протягивают со скоростью 0,2 л/мин. через систему поглотителей, содержащих по 1 мл поглотительного раствора. Отмечают уровень жидкости.
Отбор проводят непрерывно в течение суток, периодически добавляя воду до первоначального уровня.
Допустимо проводить отбор 6 — 12 раз с перерывами в 4 или 2 ч в одну и ту же систему поглотителей.
Ход анализа
Компенсатор йода отъединяют. Содержимое первого поглотительного прибора переносят в центрифужную пробирку через внутреннюю трубку компенсатора <1>. Промывают поглотительный прибор 1 мл воды и сливают ее в пробирку также через внутреннюю трубку компенсатора.
———————————
<1> Можно использовать пробирки, показанные на рис. 20.
Содержимое второго поглотительного прибора и 1 мл промывной воды переносят во вторую центрифужную пробирку.
В центрифужные пробирки, содержащие по 2 мл пробы, вносят одну каплю 0,1 н. раствора сульфита натрия для связывания йода. К бесцветному раствору пробы добавляют по 0,1 мл смеси хлорида меди и сульфита, содержимое пробирок перемешивают. Оставляют пробирки в покое на 10 — 15 мин. до появления взвеси. Далее центрифугируют в течение 5 мин. для осаждения полученной взвеси на дно пробирки и получения маленького очерченного круга.
Диаметр круга осадка в случае необходимости уменьшают вращательными движениями пробирки. Колориметрирование проводят по сравнению окраски кружков осадка с одновременно приготовленной шкалой (табл. 33). Сравнение удобно проводить при помощи зеркала, поставленного под углом 45° по отношению вертикально стоящих пробирок с пробами и стандартной шкалой.
Таблица 33
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
|
Рабочий стандартный раствор, мл |
0 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
|
Поглотительный раствор, мл |
1,0 |
0,8 |
0,6 |
0,4 |
0,2 |
0 |
|
Вода, мл |
1,0 |
1,0 |
1,0 |
1,0 |
1,0 |
1,0 |
|
Содержание ртути, мкг |
0 |
0,02 |
0,04 |
0,06 |
0,08 |
0,1 |
Во все пробирки шкалы добавляют реактивы аналогично пробе.
Расчет см. выше.
СВИНЕЦ
Мягкий металл серебристого цвета.
Плотность 11,34. Температура плавления 327,3 °C. При нагревании расплавленного свинца до 400 — 500 °C в воздухе появляются пары, быстро окисляющиеся до окиси свинца.
Температура кипения 1751 °C.
Растворим в разбавленной азотной, концентрированной серной, уксусной кислотах. В воде незначительно растворим в присутствии CO2, NH3.
Известны окисные соединения свинца: PbO — глет или массикот, Pb3O4 — сурик, PbO2 — двуокись, Pb2O — закись и многочисленные его соединения.
Токсическое действие свинца и его соединений сходно.
Свинец обладает общеядовитым действием. Наблюдаются изменения в нервной системе, крови, сосудах. Обладает кумулятивным свойством. Различная токсичность соединений свинца объясняется степенью их растворимости.
Предельно допустимая концентрация свинца и его соединений (кроме тетраэтилсвинца) в пересчете на свинец: среднесуточная — 0,0007 мг/м3.
Предельно допустимая концентрация свинца сернистого: среднесуточная 0,0017 мг/м3.
ОПРЕДЕЛЕНИЕ СВИНЦА С ХРОМАТОМ КАЛИЯ [7]
Принцип определения
Метод может быть использован для определения окиси, двуокиси, сульфата, сульфита, сульфида, хлорида, ацетата, нитрата свинца, сурика, галенита и др.
При взаимодействии свинца с хроматом калия происходит образование хромата свинца, который определяют по степени помутнения пробы.
Чувствительность определения 2 мкг в анализируемом объеме раствора.
Соли бария мешают определению.
Реактивы
1. Стандартный раствор свинца. Исходный раствор с содержанием 1 мг/мл готовят растворением 1,5884 г перекристаллизованного нитрата свинца в мерной колбе емкостью 1 л в 3% растворе ацетата аммония или буферном растворе.
Разбавляя исходный раствор в 10 раз, получают стандартный раствор А с содержанием 100 мкг/мл. Рабочий стандартный раствор Б с содержанием 10 мкг/мл готовят разбавлением раствора А в 10 раз. Растворы А и Б готовят перед употреблением.
2. Уксусная кислота, 2 и 0,5% растворы.
3. Метиловый красный. 0,1 г метилового красного растворяют в 30 мл этанола и добавляют 50 мл воды.
4. Ацетат аммония, 3% раствор, подкисленный уксусной кислотой до pH 6,6 — 6,8. pH среды устанавливают по изменению окраски метилового красного или при помощи pH-метра.
5. Ацетат натрия, 1% раствор.
6. Этанол.
7. Буферная смесь 1% раствор ацетата натрия в 0,5% растворе уксусной кислоты.
8. Серная кислота, уд. вес 1,82 — 1,84, разбавленная 1:2.
9. Азотная кислота, уд. вес 1,4, разбавленная 1:2.
10. Смесь разбавленных кислот азотной и серной 1:5.
11. Хромат калия, 1% раствор.
Отбор пробы
А. Для определения разовой концентрации воздух со скоростью до 100 л/мин. протягивают через фильтр АФА, укрепленный в патроне.
Продолжительность отбора 30 мин. и более.
Б. Для определения среднесуточной концентрации воздух со скоростью 5 л/мин. протягивают через фильтр, укрепленный в патроне, непрерывно в течение суток.
Допустимо среднесуточный отбор проводить в один и тот же фильтр со скоростью 5 — 10 л/мин. с перерывами в 2 или 4 ч.
Ход анализа
Фильтр извлекают из патрона, обрезают края, переносят в фарфоровую чашку или тигель, смачивают 1 — 2 мл смеси кислот, нагревают на песчаной бане до образования твердого остатка. Пробу переносят в муфельную печь, прикрывая крышкой. Озоление проводят при температуре не выше 600 °C, поднимать температуру выше указанной не рекомендуется во избежание улетучивания сернокислого свинца. По озолении фарфоровую чашку (при закрытой крышке) вынимают из муфельной печи, охлаждают и вносят в нее 1 мл 3% раствора ацетата аммония или буферной смеси. Раствор фильтруют через воронку диаметром 2 — 3 см с оттянутым концом, в который вложен тампон, состоящий из волокон бумажного фильтра, уложенных длиной около 0,5 см. Чашку обмывают по 0,5 мл раствором ацетата аммония или буферной смесью. Растворы фильтруют и объем доводят до 2 мл, обмывая чашку и фильтруя сливы через ту же коронку. Можно сразу в чашку внести 2 мл смеси, провести растворение сульфата свинца и перенести раствор в центрифужную пробирку, обмыть чашку, общий объем довести до 3 мл. После центрифугирования для анализа брать 2 мл прозрачного раствора. В пробу вносят 0,1 мл 1% раствора хромата калия, перемешивают и через 20 — 30 мин. сравнивают интенсивность помутнения пробы со шкалой. Рассматривают на темном фоне. Одновременно с пробами готовят стандартную шкалу и контроль. Для этого на фильтры, употребляемые для отбора проб, наносят различные количества стандартного раствора Б (табл. 34).
Таблица 34
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
|
Рабочий стандартный раствор, мл (Б) |
0 |
0,2 |
0,3 |
0,4 |
0,5 |
0,6 |
|
Содержание свинца, мкг |
0 |
2 |
3 |
4 |
5 |
6 |
Контрольный фильтр и фильтры, содержащие различные количества свинца, помещают в чашки и обрабатывают аналогично пробам.
Расчет см. выше.
ОПРЕДЕЛЕНИЕ СВИНЦА С ДИТИЗОНОМ [5]
Принцип определения
При взаимодействии неорганических соединений свинца с дитизоном образуется дитизонат, окрашенный в красный цвет, растворимый в хлороформе и четыреххлористом углероде.
Чувствительность определения 2 мкг в анализируемом объеме. Железо в количествах до 100 мкг, медь 50 мкг, цинк до 500 мкг не мешают определению.
Реактивы <1>
———————————
<1> Все реактивы должны быть приготовлены на бидистиллированной воде.
1. Стандартный раствор свинца. Исходный раствор с содержанием 1 мг/мл готовят путем растворения 1,5984 г перекристаллизованного нитрата свинца в мерной колбе емкостью 1 л в 1% растворе азотной кислоты. Рабочий стандартный раствор с содержанием 10 мкг/мл готовят разбавлением исходного в 100 раз.
2. Азотная кислота, уд. вес 1,4, х.ч., свободная от свинца, разбавленная 1:1; 2 н. раствор, последний готовят, разбавляя с 145 мл азотной кислоты, уд. вес 1,4 до 1 л.
Азотная кислота, 1% раствор; 15 мл концентрированной азотной кислоты разбавляют водой до 1 л.
3. Соляная кислота, уд. вес. 1,19, х.ч., не содержащая свинца, бидистиллированная, 1 н. раствор.
4. Винная кислота, х.ч., 10% раствор.
5. Виннокислый калий-натрий, х.ч.
6. Аммиак, уд. вес 0,91, х. ч., свободный от свинца, 1% раствор, 0,02 н. раствор.
7. Цианид калия, х.ч.
8. Сульфит натрия.
9. Буферный раствор: в 350 мл аммиака растворяют 10 г цианида калия, 15 г сульфита натрия и 15 г виннокислого калия-натрия.
10. Хлорид кальция безводный.
11. Хлороформ или четыреххлористый углерод. Очищают промыванием бидистиллированной водой, затем разбавленной соляной кислотой, 0,02 н. раствором аммиака. Осушают безводным хлоридом кальция и через 15 ч отгоняют. Аналогично регенерируют употребленный растворитель.
12. Дитизон. 30 мл дитизона растворяют в 1 л хлороформа или четыреххлористого углерода. Раствор дитизона должен быть приготовлен за 2 дня перед применением, его хранят в темном месте при охлаждении. Перед применением проверяют чистоту раствора дитизона. В пробирку вносят 5 мл раствора дитизона, промывают 5 мл 1% раствора аммиака, сильно встряхивая в течение 1 мин. Раствор применим, если слой органического растворителя бесцветный или окрашен в слабо-желтый цвет. Если раствор дитизона загрязнен продуктами окисления, то слой органического растворителя окрашен в желтый цвет и его подвергают очистке. Для этого 40 мг дитизона растворяют в 250 мл хлороформа или четыреххлористого углерода, переносят в делительную воронку емкостью 1 л, встряхивают в течение 15 мин. Далее добавляют 500 мл 0,02 н. раствора аммиака и вновь встряхивают в течение 5 — 10 мин. Оставляют в покое до расслоения жидкостей. Дитизон переходит в водный слой, а продукты окисления остаются в слое органического растворителя. Слой органического растворителя отделяют, а аммиачный слой несколько раз промывают растворителем, добавляя его по 10 — 12 мл. После очистки аммиачный слой должен иметь чисто зеленую окраску. Затем к аммиачному слою добавляют 125 мл органического растворителя и 12 мл 1 н. раствора соляной кислоты, встряхивают до тех пор, пока дитизон не перейдет в слой органического растворителя. Отделяют органический слой, содержащий дитизон, и промывают его бидистиллированной водой; затем доводят растворителем до 1 л и добавляют 5 мл воды, насыщенной сернистым ангидридом. Раствор хранят в стеклянной таре из темного стекла при +4 °C. Раствор устойчив в течение нескольких месяцев.
Применяемая посуда (делительные воронки, мерные колбы и др.) должна быть приготовлена из твердого химического стекла (типа Ена). Перед употреблением ее следует тщательно промыть горячим концентрированным раствором щелочи, горячей азотной кислотой, бидистиллированной водой до нейтральной реакции.
Фарфоровые чашки также необходимо предварительно промыть, как и стеклянную посуду, и проверить их на отсутствие свинца.
Отбор пробы см. выше.
Ход анализа
Фильтр извлекают из патрона, обрезают края, помещают в маленькую фарфоровую чашку, добавляют 10 мл азотной кислоты 1:1 и нагревают на кипящей водяной бане в течение 10 мин. При наличии в пробе PbO2 и Pb3O4 добавляют еще 10 мл концентрированной соляной кислоты и снова нагревают в течение 10 мин. Раствор отфильтровывают. Фильтр промывают 2 раза по 5 мл горячей 2 н. азотной кислотой, а затем горячей водой. Фильтрат упаривают в фарфоровой чашке на водяной бане. Сухой остаток растворяют в 5 мл горячей 10% винной кислоты и 2 мл 2 н. азотной кислоты. Раствор переносят в мерную колбу емкостью 25 мл, чашку несколько раз обмывают горячей водой. После охлаждения объем доводят до метки бидистиллированной водой.
В чистую сухую делительную воронку вносят 12,5 мл пробы, добавляют 37,5 мл буферного раствора и 12,5 мл раствора дитизона. Содержимое воронки встряхивают в течение 3 мин. После расслоения жидкостей конец делительной воронки высушивают, промывают путем спускания небольшого количества пробы. Пробу переносят в кювету и фотометрируют при длине волны 520 нм (зеленый светофильтр), кювета имеет толщину слоя 2 или 1 см по сравнению с контролем (бледно-зеленым раствором).
Содержание свинца в анализируемом объеме устанавливают по предварительно построенному калибровочному графику.
Для построения калибровочного графика готовят шкалу стандартов, при этом рабочий стандартный раствор наносят на фильтры, употребляемые для отбора. Одновременно готовят и контроль, как указано в табл. 35.
Таблица 35
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
|
Рабочий стандартный раствор, мл |
0 |
0,2 |
0,3 |
0,4 |
0,6 |
0,8 |
1,0 |
|
Содержание свинца, мкг |
0 |
2 |
3 |
4 |
6 |
8 |
10 |
Контрольный фильтр и фильтры, содержащие различные количества свинца, помещают в фарфоровые чашки и обрабатывают аналогично пробам.
Расчет см. выше.
ЛИТЕРАТУРА
1. Кузьмичева М.Н. К вопросу определения ванадия в воздухе. Гиг. и сан., 1966, 2.
2. Крылова Н.А. Определение цинка в воздухе с сульфарсазеном. Гиг. и сан., 1970, 3.
3. Логинов М.Е. Определение хрома. В кн.: М.В. Алексеева. Определение атмосферных загрязнений. М., «Медгиз», 1963.
4. Маркина Н.А. Хроматографическое разделение микроколичеств меди, кобальта, никеля в присутствии железа в атмосферном воздухе. Гиг. и сан., 1971, 9, 11.
5. Определение неорганических соединений свинца по реакции с дитизоном. Определение соединений двух- и трехвалентного хрома с дифенилкарбазидом. В сб. СЭВ: Унифицированные методы определения атмосферных загрязнений. М., 1970.
6. Полежаев Н.Г. К методике определения ртути в атмосферном воздухе. Гиг. и сан., 1956, 6.
7. Хрусталева В.А. Определение органического и неорганического свинца в воздухе гаражей. Гиг. и сан., 1952, 6.
8. Хрусталева В.А. Определение аэрозолей двуокиси марганца в воздухе производственных помещений. Гиг. и сан., 1951, 10.
Глава VII
ОКИСЬ УГЛЕРОДА, БЕНЗИН, УГЛЕВОДОРОДЫ, СИНИЛЬНАЯ КИСЛОТА
ОКИСЬ УГЛЕРОДА
Бесцветный газ, без запаха и вкуса. Плотность по отношению к воздуху 0,967. Температура кипения — 190 °C. Коэффициент растворимости в воде 0,2489 (20°), 0,02218 (30°), 0,02081 (38°), 0,02035 (40°). Вес 1 л газа при 0 °C и 760 мм рт. ст. 1,25 г. Входит в состав различных газовых смесей, коксового, сланцевого, водяного, древесного, доменного газов, выхлопных газов автотранспорта и др.
Горит синеватым пламенем. Смесь, состоящая из 1 объема кислорода и 2 объемов окиси углерода, взрывается при внесении пламени. Окись углерода является продуктом неполного сгорания веществ, в которых содержится углерод.
Обладает восстанавливающими свойствами, способна к реакциям присоединения; активность ее повышается в присутствии катализаторов.
Окись углерода токсична, вытесняет кислород из гемоглобина крови, образуя при этом карбоксигемоглобин; оказывает непосредственное действие на клетки, при этом уменьшается потребление кислорода тканями; влияет на углеводный обмен, обмен фосфора, действует на центральную нервную систему. Действие окиси углерода выражается в потере сознания, судорогах, одышке, удушье.
Предельно допустимые концентрации: максимальная разовая — 3 мг/м3, среднесуточная — 1 мг/м3.
ОПРЕДЕЛЕНИЕ ОКИСИ УГЛЕРОДА С ЙОДНОВАТЫМ АНГИДРИДОМ [2]
Принцип определения
При взаимодействии окиси углерода с йодноватым ангидридом при 130 — 150 °C происходит окисление ее до углекислоты. Последняя улавливается раствором гидрата окиси бария, избыток которого титруется соляной кислотой.
Чувствительность метода: при титровании 0,01 н. раствором соляной кислоты — 0,0014 мг (1,4 мкг), при титровании 0,005 н. раствором соляной кислоты — 0,0007 мг (0,7 мкг).
Аппаратура
1. Прибор для определения окиси углерода (рис. 21). Прибор смонтирован на деревянной основе и состоит из ряда очистительных колонок и колонки с йодноватым ангидридом, соединенных последовательно. На обратной стороне прибора расположены две очистительные колонки (9), одна наполнена пемзой, которая пропитана концентрированной серной кислотой для поглощения влаги, вторая — кусочками едкого натра, поглощающего углекислоту. На лицевой стороне прибора укреплены манометр (4) и V-образные трубки (5) высотой около 20 см и диаметром 1 см. V-образные трубки соединены между собой при помощи небольших дугообразных трубок (3).
Для очистки исследуемого воздуха от углекислоты первые две V-образные трубки заполнены кусочками едкого натра в смеси с натронной известью в соотношении 3:1. Последующие заполнены силикагелем, поглощающим предельные и непредельные углеводороды, а последняя трубка также содержит кусочки едкого натра. Верхние части всех трубок на расстоянии 2 — 5 см заполнены стеклянной ватой.
Окисление окиси углерода до углекислоты происходит в V-образной трубке (6), наполненной на 3/4 объема йодноватым ангидридом и помещенной в электрическую печь (1). Электрическая печь изготовлена из шамотной глины в смеси с волокнистым асбестом, внутри ее проложена обмотка из нихромовой проволоки, обеспечивающая обогрев печи до 200°. Контроль за температурой осуществляется при помощи реостата (2). В печи при температуре 140 — 150 °C, измеряемой термометром (8), при действии йодноватого ангидрида происходит окисление углерода до углекислоты. Выделяющийся при этом йод улавливают кристаллическим йодидом калия, который помещен в поглотительном сосуде (7). Этот сосуд, снабженный каучуком (10) и зажимом (11), защищают от действия света. Йодид калия по мере пожелтения заменяют свежим. Для поглощения йода можно применять и опилки электролитической меди. Прибор должен быть герметичен.
2. Поглотительные сосуды (см. рис. 21, А).
3. Подставки к поглотительным сосудам (см. рис. 21, Б).
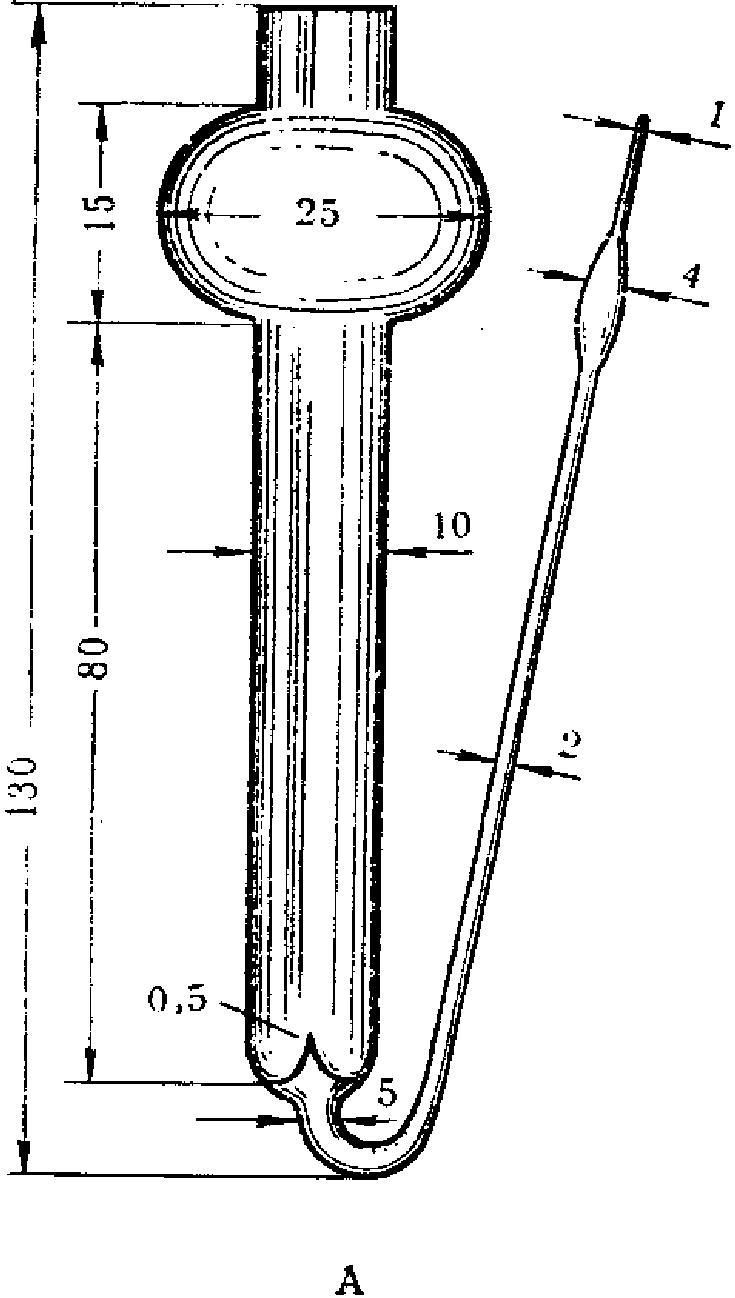
Рис. 21. Прибор для определения окиси углерода с йодноватым
ангидридом и отдельные детали к нему А — поглотительный
сосуд; Б — подставка к поглотительным сосудам; В — установка
для наполнения поглотительных сосудов; Г — пневматическая
бюретка; Д — очистительная система; Е — общий вид
4. Аспиратор, составленный из бутылей емкостью 5 — 6 л.
5. Установка для наполнения поглотительных сосудов гидратом окиси бария. Состоит из бутыли емкостью 2 — 3 л для раствора гидрата окиси бария, наполнительной бюретки емкостью 2 мл с делениями 0,01 мл. Верхний конец бюретки защищен трубкой с натронной известью (см. рис. 21, В). К установке приложены две очистительные колонки (см. рис. 21, Д), наполненные натронной известью в смеси с кусочками едкого натра, и пневматическая микробюретка (см. рис. 21, Г). Последняя представляет собой пипетку емкостью 2 мл с делением 0,01 мл. Нижний ее конец оттянут в капилляр диаметром не более 0,1 см. Верхняя часть микробюретки соединена с каучуковой трубкой длиной около 50 см, которая в свою очередь соединена с резиновым баллоном емкостью 20 — 30 мл, помещенным между пластинкой микрометрического винта и деревянной подставкой. При погружении кончика микробюретки в жидкость и расширении баллона проводят засос жидкости в бюретку. Обратное движение винта способствует вытеснению жидкости из микробюретки.
6. Бутыли для отбора проб воздуха емкостью 2 — 3 л, в которые вставлены пробки с двумя стеклянными трубками. Одна из них доходит почти до дна, другая — короткая. На верхние концы трубок надеты каучуковые трубки, закрытые стеклянными заглушками и винтовыми зажимами (см. рис. 21) (12, 13, 14, 15).
В настоящее время завод «Химлаборпосуда» изготавливает прибор ПОУ для определения окиси углерода по МРТУ 42-2068-20.
Реактивы
1. Соляная кислота 0,01 н. раствор, 0,005 н. раствор (точно приготовленный из фиксанала).
2. Гидрат окиси бария, 0,01 и 0,005 н. раствор. Готовят растворением 4,5 г гидрата окиси бария (кристаллы должны быть прозрачны, без налета углекислого бария) и 5 г хлорида бария в колбе емкостью 2 л. Колбу закрывают пробкой, в которую вмонтированы две стеклянные трубки, одна доходит почти до дна, другая короткая. Концы трубок защищают от влияния углекислоты натронной известью. Содержимое колбы взбалтывают и оставляют в покое на 1 — 2 суток, до получения прозрачного раствора. Прозрачный раствор из колбы сифонируют в бутыль емкостью 2 — 3 л, которую предварительно и в момент сифонирования продувают током воздуха, лишенным углекислоты. Бутыль с раствором гидрата окиси бария закрывают пробкой, в которую вмонтированы две трубки — длинная и короткая. Короткую трубку защищают натронной известью в смеси с кусочками едкого натра.
От влияния углекислоты длинную трубку соединяют с бюреткой. Верхняя часть бюретки также защищена натронной известью.
3. Хлорид бария.
4. Фенолфталеин, 0,5% раствор в 50% растворе этанола.
5. Этанол.
6. Натронная известь гранулированная.
7. Едкий натр, 40% раствор.
8. Пемза гранулированная.
9. Хлорид натрия, 26% раствор.
10. Силикагель, размер зерен 2 — 4 мм, активированный при 200 °C в течение 30 — 40 мин.
11. Серная кислота, уд. вес 1,84.
12. Йодид калия.
13. Йодноватый ангидрид, гранулированный. Порошкообразный йодноватый ангидрид или йодноватую кислоту помещают в фарфоровую чашку, увлажняют водой. Чашку ставят на водяную баню и по мере испарения воды, помешивают стеклянной палочкой. Образующиеся гранулы доводят до размера 2 — 3 см, подсушивают в сушильном шкафу при 105 °C в течение 1 — 2 ч. V-образную трубку наполняют 8 — 10 г гранулированного йодноватого ангидрида, помещают ее в электрическую печь прибора, соединяют с очистительной системой и продувают воздухом в течение нескольких часов. Печь нагревают, постепенно повышая температуру. Когда выделение влаги и йода прекратится, температуру повышают до 200° и продувают еще в течение 1 1/2 — 2 ч. Йодноватый ангидрид сохраняет восстановительную способность в течение длительного времени; 1 г йодноватого ангидрида способен окислить 400 г окиси углерода. Окислительную способность йодноватого ангидрида проверяют на пробах воздуха с известным содержанием окиси углерода.
14. Медь электролитическая. Электролитическую медь просеивают через сито или освобождают от пыли путем отмучивания в этиловом эфире. Эфир отделяют, опилки высушивают при комнатной температуре. Затем медь помещают в V-образную трубку, прикрывают небольшим слоем стеклянной ваты и продувают воздухом в течение 30 мин., нагретым не выше 100 °C и очищенным от углекислоты.
Отбор пробы
А. Для определения разовой концентрации воздух отбирают в бутыль емкостью 2 — 3 л, закрытую резиновой пробкой, в которую вмонтированы две стеклянные трубки — длинная и короткая. Объем бутыли заполняют 26% раствором хлорида натрия.
При отборе к длинному концу трубки присоединяют каучуковую трубку длиной 40 — 50 см и выливают раствор хлорида натрия со скоростью около 100 мл/мин. При этом исследуемый воздух поступает в бутыль через короткую трубку. Продолжительность отбора 20 — 30 мин. По окончании отбора отверстия трубок закрывают стеклянными заглушками и винтовыми зажимами. Отбор проб может быть проведен в пипетки емкостью 0,5 — 1 л.
Б. Для определения среднесуточной концентрации пробу отбирают в течение 24 ч в аспиратор емкостью 3 л, наполненный 26% раствором хлорида натрия. Скорость отбора 0,1 л/ч, через 24 ч в верхней бутыли аспиратора должно быть около 0,5 л раствора хлорида натрия.
Ход анализа
а) Подготовка прибора. Перед началом анализа устанавливают соотношение растворов гидрата окиси бария и соляной кислоты. Для этого поглотительный сосуд продувают в течение 2 мин. током очищенного воздуха, затем ставят его вертикально под кончик бюретки, которая наполнена гидратом окиси бария. В токе очищенного воздуха спускают из бюретки в сосуд 2 мл раствора и добавляют каплю фенолфталеина. Скорость поступления очищенного воздуха регулируют так, чтобы жидкость не попадала в широкую часть поглотительного сосуда. Наполнив один поглотитель, при помощи каучуковой трубки соединяют широкий конец его с капиллярной трубкой второго поглотительного сосуда. Так же в токе очищенного воздуха вносят во второй поглотитель 2 мл раствора гидрата окиси бария. Таким образом, получают два последовательно соединенных поглотительных прибора, наполненных по 2 мл раствора гидрата окиси бария, через который проходит ток очищенного воздуха. Не прекращая ток воздуха, начинают титрование со второго поглотительного сосуда. Титрование проводят раствором соляной кислоты до обесцвечивания раствора. Далее второй поглотитель отъединяют и титруют первый. На 2 мл 0,01 н. раствора гидрата окиси бария должно пойти примерно 2 мл (не более) 0,01 н. раствора соляной кислоты. Результаты титрования каждого поглотительного сосуда не должны отличаться между собой более чем на 0,01 мл соляной кислоты.
б) Контрольный анализ. Для получения воздуха, не содержащего окиси углерода, перед прибором включают систему, состоящую из хлоркальциевой трубки, наполненной хлоридом кальция, последовательно соединяют с V-образной трубкой, наполняют йодноватым ангидридом и помещают в дополнительную печь.
Дополнительную печь и печь прибора нагревают до 130 — 150 °C. Наполняют два поглотительных прибора раствором гидрата окиси бария аналогично описанному выше. Поглотительные приборы присоединяют к прибору. Через всю систему протягивают 1 л воздуха в течение часа. Поглотительные приборы с раствором гидрата окиси бария устанавливают почти в горизонтальном положении с тем, чтобы воздух проходил через раствор непрерывной цепочкой. При прохождении воздуха через первую трубку с йодноватым ангидридом имеющаяся окись углерода полностью окисляется до углекислого газа, которая далее улавливается в очистительной системе прибора. После прохождения 1 л воздуха через поглотительные приборы, объем которого регистрируется специальным цилиндром, закрывают зажим непосредственно перед прибором и наблюдают прекращение потока воздуха через поглотительные приборы.
Затем при помощи микробюретки титруют раствор гидрата окиси бария 0,01 н. раствором соляной кислоты. Титр раствора гидрата окиси бария получается более низкий, чем при непосредственном титровании раствора. Снижение обычно не превышает 0,02 мл 0,01 н. раствора соляной кислоты. Контрольное определение повторяют до получения совпадающих результатов.
в) Анализ проб проводят аналогично контрольному. Но при этом отключают дополнительную очистительную систему от прибора, служащую для получения воздуха, не содержащего окиси углерода. К прибору присоединяют емкость с пробой, пропускают через прибор 500 мл исследуемого воздуха (для заполнения системы). Наполняют и присоединяют к прибору поглотительные приборы и протягивают 1 л исследуемого воздуха в течение 50 — 60 мин. Контрольный анализ проводят перед каждым определением.
Расчет ведут по формуле:
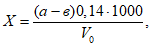
где: X — концентрация окиси углерода в миллиграммах на 1 м3 воздуха; а — количество 0,01 н. раствора соляной кислоты, израсходованное на титрование гидрата окиси бария при контрольном анализе; в — количество 0,01 н. раствора соляной кислоты, израсходованное на титрование гидрата окиси бария при анализе пробы; V0 — количество исследуемого воздуха (в литрах), приведенное к нормальным условиям; 1000 — множитель для выражения концентрации в миллиграммах на 1 м3 воздуха; 0,14 — количество окиси углерода, соответствующее 1 мл точно 0,01 н. раствору соляной кислоты (в миллиграммах); 0,007 — количество окиси углерода, соответствующее 1 мл точно 0,005 н. раствора соляной кислоты.
ОПРЕДЕЛЕНИЕ ОКИСИ УГЛЕРОДА НА ГАЗОАНАЛИЗАТОРЕ [3]
Принцип определения
При сжигании окиси углерода на платиновой спирали, нагретой до температуры красного каления (600°), образуется углекислота, которую поглощают раствором гидрата окиси бария. Количество углекислоты определяют по разности титрования избытка гидрата окиси бария раствором соляной кислоты до сжигания пробы и после него.
Чувствительность определения при титровании 0,01 н. раствором соляной кислоты 0,0014 мг (1,4 мкг).
При титровании 0,005 н. раствором соляной кислоты 0,0007 мг (0,7 мкг).
Определению мешает метан.
Газоанализатор состоит из очистительной и аналитической систем. Схема анализатора приведена на рис. 22.
Рис. 22. Схема титрометрического газоанализатора
Очистительная система предназначается для поглощения углеводородов и двуокиси углерода из воздуха. Она состоит из колонки предварительного сожжения органических веществ воздуха (1), двух змеевиков-барботеров (2, 3), заполненных 40% раствором щелочи, патрона с хлоридом кальция (4а), двух V-образных трубок (4б, 4в); трубка 4б наполнена пемзой, смоченной серной кислотой удельного веса 1,82 — 1,84; трубка 4в — силикагелем.
В колонку предварительного сожжения впаяна спираль из платиновой проволоки длиной 70 мм и сечением 0,3 мм. Колонка обеспечивает полное сожжение горючих примесей в воздухе до двуокиси углерода, поглощаемой в барботерах с 40% раствором щелочи. Каждый из барботеров представляет собой сосуд, в который впаян змеевик.
В аналитическую часть входят следующие детали:
1. Колонка для сожжения окиси углерода по конструкции не отличается от колонки очистительной системы. Назначение колонки — каталитическое сожжение окиси углерода до двуокиси углерода.
2. Поглотительный сосуд-змеевик (6) — наиболее ответственная деталь аналитической части. Служит для поглощения двуокиси углерода раствором едкого бария.
3. Аспиратор, трехходовой кран и уравнительная склянка (16, 17), служащие для засасывания пробы воздуха в прибор, а также для замера его количества. Аспиратор представляет собой цилиндрический сосуд, в центре которого находится трубка, доходящая почти до дна нижней узкой части аспиратора. Тубус аспиратора соединяется посредством резиновой трубки с уравнительной склянкой, заполненной водой. Ниже тубуса в суженной части аспиратора находится слой ртути, препятствующий попаданию воды в прибор при вытеснении воздуха из аспиратора. В верхней, суженной, части аспиратора имеется отвод для удаления проанализированного воздуха из прибора через ртутный затвор.
4. Две микробюретки для титрования растворов едкого бария и соляной кислоты (13, 14) емкостью 4,5 мл с делениями 0,01 мл. В каждую микробюретку напаян сверху полый стеклянный шар, имеющий два отвода. Один отвод шара служит для соединения бюретки с атмосферой через защитный барботер и четырехходовой кран. Другой отвод шара соединен со сливной склянкой и служит для сливания в нее избыточной жидкости.
5. Сливная склянка (12) служит для сливания отработанного раствора из поглотительного сосуда и избытка раствора из микробюреток через трехходовой кран (10).
6. Краны трехходовые (7), позволяющие спускать растворы Ba(OH)2 и HCl в поглотительный сосуд (6), а также для заполнения бюреток растворами. Все краны смазывают полувакуумной смазкой.
7. Четырехходовой кран (11), к которому присоединяется резиновый баллон (19). С помощью резинового баллона создается разрежение в приборе.
8. Для склянки (15) с титрованными растворами. Пробку на склянке с едким барием герметизируют парафином или замазкой.
9. Трансформатор на 2 — 6 В (на рисунке не помечен).
10. Пипетка газовая емкостью 500 мл или бутыль с сифоном объемом не менее 1 л.
11. Защитные барботеры (20, 21).
12. Ртутный затвор (22).
Реактивы
1. Ртуть.
2. Раствор для поглощения паров ртути. Смешивают 30 г йодистого калия и 2,5 г кристаллического йода. В смесь постепенно вливают 1 л дистиллированной воды.
Растворы соляной кислоты, гидрата окиси бария, едкого кали, хлористого натрия и др. готовят так, как указано при определении окиси углерода с йодноватым ангидридом.
Отбор пробы
а) При определении максимально разовой концентрации отбирают две пробы в газовые пипетки емкостью 400 — 500 мл, наполненные 26% раствором поваренной соли. В месте отбора раствор выпускают из пипетки в течение 5 — 10 мин. и таким образом заполняют их исследуемым воздухом. По окончании отбора закрывают зажимы на пипетке, а в концы резиновых трубок вставляют стеклянные палочки.
б) Для определения среднесуточной концентрации отбирают пробу в течение 24 ч в аспиратор емкостью 3 л, наполненный 26% раствором поваренной соли. Скорость отбора 0,1 л/ч. Через 24 ч в верхней бутыли аспиратора должно остаться раствора около 0,5 л.
Подготовка прибора для производства анализа
Для приведения прибора в рабочее состояние необходимо сделать следующие операции:
1. Заполнить бюретки растворами, для чего создают в них разрежение резиновым баллоном путем соединения четырехходового крана с внешним воздухом. В таком положении воздух из баллона выжимают, сжимая его рукой. Не разжимая руку, закрывают кран, соединяют баллон с бюретками и сливной склянкой. В бюретках и сливной склянке при разжимании баллона создается разрежение. Трехходовые краны провертывают таким образом, чтобы бюретки соединились со склянками, наполненными титрованными растворами. При этом бюретки заполняются растворами.
2. Заполнить поглотительный сосуд-змеевик в токе воздуха титрованными растворами. Аспиратор заполняют водой, из аспиратора медленно сливают воду в уравнивательную склянку. Вращая четырехходовой кран, создают в бюретках атмосферное давление. Краны (7) поочередно поворачивают в сторону поглотительного сосуда, соединяя его с бюретками и заполняя их то одним титрованным раствором, то другим, так, чтобы верхние капилляры сосуда погрузились в раствор. Создавая разрежение в бюретках с помощью баллончика (19) и четырехходового крана, засасывают из поглотительного сосуда по капиллярам раствор из бюретки. Если в процессе работы образуются пузырьки в капиллярах и отводах, то их вытесняют так же. По удалении пузырьков воздуха жидкость из поглотительного сосуда выливают в сливную склянку в токе воздуха. Это делают с помощью баллона и четырехходового крана, открывая кран у поглотительного сосуда в сторону сливной склянки, при этом жидкость выливается.
Чтобы вылить жидкость из сливной склянки, следует баллон наполнить воздухом, снять заглушку с резиновой трубки на сливной склянке, сжать баллон рукой, и жидкость из склянки выльется.
После этого необходимо произвести проверку прибора на герметичность. Для этого в поглотительный сосуд спускают растворы едкого бария и соляной кислоты, закрывают заглушкой входное отверстие прибора, наполняют аспиратор водой, спускают уравнительную склянку в нижнее положение, открывают кран на полный ход и включают колонки для сожжения. В таком положении прибор оставляют на 5 мин. Если прибор герметичен, в отдельных его частях не наблюдается движения жидкости. Если прибор не герметичен, то следует тщательно осмотреть его, чтобы установить, в какой части имеется негерметичность. В случае обнаружения негерметичности следует сменить резиновые трубки и снова проверить прибор на герметичность. После этой операции прибор готов для проведения анализа.
Контрольный анализ
Для определения титра раствора едкого бария включают колонку (1) в очистительной системе. Заполняют аспиратор водой, опускают уравнительную склянку вниз, заполняют бюретки растворами, создавая разрежение в бюретках, как описано выше. Затем выравнивают давление в бюретках с помощью четырехходового крана, наливают в поглотительный сосуд 5 — 6 мл раствора едкого бария (точно) в зависимости от размера барботера и пропускают 500 мл воздуха. Не прекращая тока воздуха, проводят титрование раствора едкого бария 0,01 н. раствором соляной кислоты до обесцвечивания раствора.
На контрольный анализ и анализ пробы берут одинаковое количество раствора едкого бария. Контрольный анализ проводят несколько раз до получения совпадающих результатов.
Ход анализа
В поглотительный сосуд наливают 4 — 4,5 мл раствора едкого бария в токе воздуха при включенной колонке в очистительной системе. Присоединяют газовую пипетку с пробой к прибору таким образом, чтобы верхний конец пипетки, закрытый зажимом, был соединен с колонкой предварительного сожжения, а нижний, тоже закрытый зажимом, — с уравнительным сосудом, наполненным водой. Открывая нижний зажим пипетки, создают давление в ней.
Аспиратор заполняют водой до метки «0». Уравнительную склянку аспиратора опускают вниз. Выключают колонку в очистительной системе и включают колонку в аналитической системе. Верхний зажим пипетки с пробой открывают и с помощью аспиратора протягивают анализируемый воздух из пипетки через прибор со скоростью 17 — 20 мл/мин. Отсчет анализируемого воздуха ведут по аспиратору прибора.
Рис. 23. Титрометрический газоанализатор-ГТУ, изготовленный
по МРТУ 42-2067-02 (Б), А — вид сбоку; Б — общий вид
Анализируемый воздух поступает в колонку очистительной системы (не включенную в сеть) в барботеры со щелочью для освобождения от углекислоты, затем в хлоркальциевую трубку (4,а) и в трубки 4,б, 4,в для осушки и очистки воздуха от углеводородов. После очистительной системы воздух поступает в колонку (5), где окись углерода сгорает до углекислоты на раскаленной платиновой спирали.
Образовавшаяся углекислота поступает в поглотительный сосуд, наполненный раствором едкого бария. После сжигания пробы пипетку отъединяют, затем пропускают 100 мл «чистого» воздуха для проталкивания анализируемого воздуха в аналитическую часть, выключая колонку в аналитической системе, включая колонку в очистительной системе, и проводят титрование раствора в поглотительном сосуде в токе воздуха. По разности результатов титрования до и после анализа вычисляют содержание окиси углерода. 1 мл 0,01 н. раствора соляной кислоты соответствует 0,14 мг окиси углерода (полученную величину умножают на 0,14).
Расчет анализа см. выше.
Примечание. Газоанализатор титрометрический: изготавливается по МРТУ 42-2067-62. Изготовитель — завод «Лаборприбор» (рис. 23).
БЕНЗИН
Смесь углеводородов ряда метана, непредельных, ароматических углеводородов, нафтенов.
Существуют нефтяные, сланцевые, синтетические и другие бензины.
Нефтяные бензины — продукты, получаемые в процессе перегонки нефти.
Сланцевый бензин — продукт пиролиза горючих сланцев.
Синтетические бензины — продукты гидрирования окиси углерода, ацетилена и др. и дальнейшей полимеризации.
Бензины представляют собой легко испаряющиеся жидкости, без цвета или с желтоватым оттенком.
Химический состав различных бензинов отличается и зависит от способов их получения. В состав сланцевых бензинов входят сернистые органические соединения.
Бензины обладают наркотическим действием, оказывают влияние на центральную нервную систему. Вызывают головную боль, раздражение слизистых оболочек, дерматиты.
Предельно допустимые концентрации (мг/м3):
|
Максимальная разовая |
Среднесуточная |
|
|
Бензин (нефтяной, малосернистый в пересчете на углерод) |
5 |
1,5 |
|
Бензин сланцевый (в пересчете на углерод) |
0,05 |
0,05 |
ОПРЕДЕЛЕНИЕ БЕНЗИНА (СУММЫ УГЛЕВОДОРОДОВ)
НА ГАЗОАНАЛИЗАТОРЕ [4]
Принцип определения
При каталитическом сожжении углеводородов (бензина, спирта и других легких углеводородов) происходит образование углекислого газа, который поглощается раствором гидрата окиси бария. Его избыток оттитровывается раствором соляной кислоты. Концентрацию суммы углеводородов пересчитывают на углерод (C). Мешают определению органические соединения, которые поглощаются щелочью, кислотой или реагируют с ними (альдегиды, кетоны, фенолы, сложные эфиры), а также галогеноорганические соединения.
Чувствительность определения в пересчете на углерод при титровании 0,01 н. раствором соляной кислоты 0,0006 мг (0,6 мкг), при титровании 0,005 н. раствором соляной кислоты 0,0003 мг (0,3 мкг).
Метан мешает определению.
Прибор, реактивы, отбор проб, подготовка прибора к анализу и определение титра гидрата окиси бария описаны при определении окиси углерода на газоанализаторе (см. выше). При анализе пробы на содержание бензина V-образные трубки, наполненные силикагелем и пемзой, пропитанной серной кислотой (поглощающие углеводороды), отключают от прибора.
ОПРЕДЕЛЕНИЕ БЕНЗИНА (СУММЫ УГЛЕВОДОРОДОВ)
В ПРИСУТСТВИИ ОКИСИ УГЛЕРОДА НА ГАЗОАНАЛИЗАТОРЕ
Принцип определения
Метод основан на каталитическом сожжении бензина и окиси углерода до CO2, поглощении двуокиси углерода раствором барита и обратном титровании его избытка раствором соляной кислоты; в той же пробе по этому принципу определяется окись углерода, а затем до разности находят концентрацию бензина в пересчете на углерод.
Чувствительность определения в пересчете на углерод 0,0006 мг (0,6 мкг). Мешает определению присутствие других органических веществ. Аппаратура, реактивы, отбор пробы, подготовка прибора к анализу, определение титра едкого барита см. «Определение окиси углерода на газоанализаторе» выше.
Контрольный анализ
Через прибор в поглотительный сосуд с едким баритом при включенных колонках протягивают 500 мл «чистого» воздуха, титруют раствор барита раствором HCl, отмечая количество миллилитров HCl, пошедшее на обратное титрование гидроокиси бария (а).
Анализ пробы
Сначала определяют общее содержание углеводородов и окиси углерода в исследуемом воздухе. При этом колонки очистительной системы (колонки с пемзой, пропитанной H2SO4 и силикагелем) отсоединяют. Силикагель используют марки СК и СМ, размер зерен 1 — 2 мм. Его предварительно активируют при 250°C. Спускают из бюретки точно такое же количество раствора едкого барита, какое было взято при проведении контрольного анализа.
Короткий конец бутыли с пробой присоединяют к прибору, другой конец сифона соединяют со склянкой, наполненной 26% раствором хлористого натрия. Открывают зажимы сифона бутыли сначала длинной трубки, проверяя герметичность бутыли (если бутыль герметична, то через короткое время прекращается поступление раствора в бутыль). Затем открывают зажим короткой трубки, и солевой раствор, поступая в бутыль с пробой воздуха, вытесняет последний в прибор.
Объем пропускаемого воздуха учитывают по аспиратору прибора. Выключают колонку предварительного сожжения и оставляют включенной колонку аналитической части, с помощью аспиратора и уравнительной склянки протягивают исследуемый воздух со скоростью 17 — 20 мл/мин. Образующаяся при сожжении бензина и окиси углерода двуокись углерода поступает в поглотительный сосуд с раствором барита. После вытеснения 500 мл исследуемого воздуха пробы из бутылки последнюю отсоединяют от прибора и присоединяют к прибору очистительную систему (колонки, заполненные пемзой, пропитанной серной кислотой и силикагелем), пропуская через нее 100 мл «чистого» воздуха для проталкивания остатка анализируемого воздуха в аналитическую часть. Затем включают колонку предварительного сожжения и пропускают еще 100 мл «чистого» воздуха, затем производится титрование избытка барита раствором HCl с отметкой количества соляной кислоты в миллилитрах.
Определение окиси углерода
Определение окиси углерода в пробе производится так же, как было указано выше, с той лишь разницей, что анализируемый воздух до поступления в барботеры предварительно очищают от бензина пропусканием через очистительную систему (колонки — одну, заполненную пемзой, пропитанной серной кислотой, и другую, заполненную силикагелем), отмечают количество соляной кислоты в миллилитрах, пошедшее на титрование избытка барита. Концентрацию бензина в пробе воздуха в пересчете на углерод (мг/м3) рассчитывают по формуле:
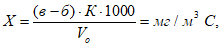
где: в — количество соляной кислоты (мл), пошедшее на титрование избытка раствора барита после поглощения CO2, образовавшегося при сожжении окиси углерода и углеводородов пробы воздуха; б — количество соляной кислоты (мл), пошедшее на титрование избытка раствора барита после поглощения CO2, образовавшегося при сожжении окиси углерода пробы воздуха; K = 0,06 — количество C (мг), соответствующее 1 мл 0,01 н. раствора HCl; V0 — количество исследуемого воздуха (л), приведенное к нормальным условиям (см. выше).
В случае необходимости вычислить и концентрацию окиси углерода в исследуемой пробе (в миллиграммах на 1 м3 воздуха) (X), ее рассчитывают по следующей формуле:
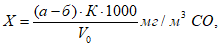
где: а — количество миллилитров HCl, пошедшее на титрование избытка раствора барита после протягивания «чистого» воздуха, при контрольном
анализе; б — количество HCl в 1 мл, пошедшее на титрование избытка раствора барита после поглощения CO2, полученного при сожжении окиси углерода из пробы воздуха; K = 0,14 — количество CO в миллиграммах, соответствующее 1 мл 0,01 н. раствора HCl; V0 — количество исследуемого воздуха пробы в литрах, приведенного к нормальным условиям (см. выше).
ЦИАНИСТЫЙ ВОДОРОД (СИНИЛЬНАЯ КИСЛОТА)
Бесцветная, легко подвижная, весьма летучая жидкость с запахом горького миндаля. Хорошо растворима в воде, органических растворителях. Плотность паров по отношению к воздуху 0,93. Температура кипения 25,5 °C, температура замерзания — 14 °C. Упругость паров 647,9 мм рт. ст. при 21,4 °C. Цианистый водород относится к сильно токсическим соединениям, вызывает параличи дыхательных путей, проникает через кожные покровы. Среднесуточная предельно допустимая концентрация 0,01 мг/м3.
ОПРЕДЕЛЕНИЕ ЦИАНИСТОГО ВОДОРОДА
ПО ОБРАЗОВАНИЮ ПОЛИМЕТИНОВОГО КРАСИТЕЛЯ [1]
Принцип определения
При взаимодействии цианистого водорода и пиридина с анилином образуется дианилид глютаконового альдегида, относящийся к классу полиметиновых красителей.
Чувствительность определения 0,1 мкг в анализируемом объеме раствора.
Мешают определению хлор, бром, влияние их устраняют поглощением в индикаторную трубку.
Реактивы
1. Поглотительный раствор — 0,1 н. раствор едкого натрия.
2. Исходный стандартный раствор из фиксанала 0,1 н. раствора роданида аммония, 1 мл 0,1 н. раствора которого соответствует 5,81 мг иона SCN или 2,7 мг CN.
Рабочий стандартный раствор с содержанием 1 мкг/мл готовят перед употреблением соответствующим разбавлением водой.
3. Соляная кислота, 0,1 н. раствор.
4. Едкий натр, 0,01 н. раствор.
5. Бромная вода. Смешивают 0,5 мл брома с 50 мл воды.
6. Пиридин. Очищают кипячением в течение часа с обратным холодильником над кристаллической щелочью. Затем, добавив кристаллическую щелочь, перегоняют. Отбирают фракцию, кипящую при 114 — 116°. Хранят в темной закрытой склянке.
7. Анилин. Перегоняют при 184,5 °C.
8. Уксусная кислота, ледяная, х.ч.
9. Индикаторная вата. Гигроскопическую вату промывают горячим спиртом, высушивают при 80 — 90 °C. В раствор, состоящий из 40 г йодида калия и 100 мл воды, погружают на 20 мин. 10 г ваты. Вату отжимают между фильтровальной бумагой и высушивают при 80 °C. Хранят в склянке из темного стекла.
10. Составной реактив готовят смешением 10 мл пиридина, 5 мл ледяной уксусной кислоты и 1 мл анилина.
11. Гидразинсульфат, 0,5% раствор.
Отбор пробы
Воздух со скоростью 0,2 — 0,3 л/мин. протягивают через два последовательно соединенных поглотительных прибора Зайцева, наполненных по 2 мл поглотительного раствора. При наличии хрома, брома в воздухе перед поглотительными приборами включают индикаторную трубку с ватой. Протягивают не менее 80 л исследуемого воздуха.
Ход анализа
Из каждого поглотительного прибора 1 мл пробы помещают в пробирки, прибавляют по 1 мл 0,1 н. соляной кислоты и 0,1 мл бромной воды и по каплям 0,5% раствора гидразинсульфата до обесцвечивания. Перемешивают и вносят по 1 мл составного реактива и доводят объем до 4 мл водой. Через 15 мин. фотометрируют в кюветах с толщиной слоя 1 см при длине волны 485 нм по сравнению с контролем, который готовят одновременно с пробой. Содержание синильной кислоты определяют по предварительно построенному калибровочному графику. Готовят калибровочный график (табл. 36).
Таблица 36
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
|
Рабочий стандартный раствор, мл |
0 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
1 |
|
Поглотительный раствор, мл |
1 |
0,9 |
0,8 |
0,6 |
0,4 |
0,2 |
0 |
|
Содержание синильной кислоты, мкг |
0 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
1 |
Все пробирки шкалы стандартов и пробу обрабатывают, аналогично измеряют оптические плотности и строят график. Шкала стандартов может быть использована для визуального определения.
Расчет см. выше.
ЛИТЕРАТУРА
1. Беляков А.А., Мельникова Л.В. Определение синильной кислоты. В кн.: Определение вредных веществ в воздухе производственных помещений. Волго-вятское книжное издательство. Горький, 1970.
2. Методы определения окиси углерода в воздухе. В сб.: Технические условия на методы определения вредных веществ в воздухе. М., 1964, в. III.
3. Сендерихина Д.П. Определение окиси углерода на титрометрическом газоанализаторе. В сб.: Информационно-методические материалы. Методы гигиенических исследований. Московский научно-исследовательский институт санитарии и гигиены им. Ф.Ф. Эрисмана. М., 1955, в. 5.
4. Сендерихина Д.П. Определение бензина в присутствии окиси углерода на газоанализаторе. В сб.: Методы гигиенических исследований. Информационно-методические материалы. Московский научно-исследовательский институт санитарии и гигиены им. Ф.Ф. Эрисмана. М., 1955, в. 5.
Глава VIII
УГЛЕВОДОРОДЫ АРОМАТИЧЕСКОГО РЯДА
БЕНЗОЛ
Бесцветная прозрачная жидкость с характерным запахом, плотность при 20° 0,879. Температура плавления 5,53 °C; температура кипения 80,1°. Бензол хорошо растворим в эфире, спирте, хлороформе и других органических растворителях; он является хорошим растворителем жиров, смол, масел, ряда других органических веществ; растворяет серу, фосфор и йод.
Высокие концентрации бензола оказывают влияние на центральную нервную систему, а также вызывают изменения крови и кроветворных органов.
Бензол довольно сильно раздражает кожу.
Предельно допустимые концентрации: максимальная разовая 1,5 мг/м3; среднесуточная 0,80 мг/м3.
ОПРЕДЕЛЕНИЕ БЕНЗОЛА НИТРОВАНИЕМ [1]
Принцип определения
При нитровании бензола образуется динитробензол, который в щелочной среде в присутствии кетона дает соединение, окрашенное в красно-фиолетовый цвет.
Чувствительность определения 1 мкг в анализируемом объеме раствора. Другие ароматические углеводороды мешают определению.
Реактивы
1. Поглотительный раствор — нитрационная смесь: 10 г нитрата аммония, высушенного при 80 °C, растворяют в 100 мл серной кислоты, уд. вес 1,84.
2. Стандартный раствор бензола: исходный раствор готовят в мерной колбе емкостью 50 мл, в которой помещено 1 — 2 мл нитрационной смеси. Колбу взвешивают и вносят в нее 2 — 3 капли бензола и вновь взвешивают. По разности двух взвешиваний устанавливают количество бензола в колбе. Раствор встряхивают и колбу помещают в кипящую водяную баню на 40 мин. По истечении времени раствор охлаждают и постепенно, осторожно в колбу вливают воду при помешивании раствора. Колбу в это время помещают в холодную водяную баню. Раствор доводят до метки после того, как температура его сравнится с комнатной.
Высчитывают содержание бензола в 1 мл раствора. Рабочий стандартный раствор с содержанием 100 мкг/мл готовят в мерной колбе емкостью 50 мл, в нее вливают такое количество исходного раствора, которое содержит 5 мг бензола. Раствор доводят до метки нитрационной смесью, разбавленной водой в отношении 1:3.
3. Метилэтилкетон (бутанон) бесцветный, свежеперегнанный, точка кипения 74 — 80 °C. Бутанон, бывший в употреблении, можно применять вновь, предварительно отделив от щелочи в делительной воронке, промыв водой, осушив хлоридом кальция, дважды перегнав его. Полученный бутанон не должен давать окраску со щелочью.
4. Едкий натр или едкое кали, 20% раствор.
5. Серная кислота, уд. вес 1,84.
6. Бензол, свежеперегнанный (температура кипения 80,1 °C).
7. Нитрат аммония, высушенный при 80 °C.
8. Аммиак, 25% раствор.
Отбор пробы
а) Для определения разовой концентрации воздух со скоростью 0,2 — 0,3 л/мин. протягивают через поглотительный прибор с пористым фильтром N 1 (малая модель), содержащий 2 мл поглотительного раствора. Продолжительность отбора 20 — 40 мин.
б) Для определения среднесуточной концентрации воздух в течение суток протягивают через поглотительный прибор непрерывно, со скоростью 0,1 л/мин.
Допустимо отбор проб проводить 6 — 12 раз с перерывами в 2 — 4 ч в один и тот же поглотительный прибор.
Ход анализа
Поглотительный прибор с пробой помещают в кипящую водяную баню на 40 мин. По охлаждении раствора его сливают в коническую колбу емкостью 50 мл и поглотительный прибор ополаскивают несколько раз водой, на что расходуют 6 — 8 мл. Воду приливают к пробе осторожно, избегая сильного разогревания раствора.
После охлаждения пробу нейтрализуют 25% раствором аммиака по лакмусу. Нейтрализацию проводят из бюретки с краном при охлаждении.
Затем пробу переводят в делительную воронку емкостью 25 мл, добавляют 4 мл бутанона и раствор встряхивают в течение 5 мин. Раствору дают расслоиться и водный нижний слой выливают через кран воронки, а верхний бутаноновый переливают через верх воронки в пробирку, объем раствора доводят до 4 мл бутаноном.
В пробирку с пробой наливают 1 мл 20% раствора едкого натра, содержимое пробирки встряхивают в течение нескольких минут и через 15 — 20 мин. измеряют оптическую плотность в кювете с толщиной слоя 1 см по сравнению с контролем, который готовят одновременно и аналогично пробе. Определение проводят при длине волны 574 нм. Содержание бензола в анализируемом объеме определяют по предварительно построенному калибровочному графику. Для построения калибровочного графика готовят шкалу стандартов из стандартного бутанонового раствора. Для этого 2 мл рабочего стандартного раствора вливают осторожно в коническую колбу емкостью 100 мл, в которую предварительно влито 8 — 10 мл воды. Раствор охлаждают и нейтрализуют 25% раствором аммиака в присутствии лакмуса.
Охлажденный нейтральный раствор выливают в делительную воронку и приливают к нему 10 мл бутанона. Растворы встряхивают в течение 10 мин. и оставляют в покое до полного разделения слоев. Затем нижний водный слой сливают через кран воронки, а верхний бутаноновый слой переливают через верхний край воронки в мерную колбу емкостью 10 мл. Объем раствора доводят бутаноном до метки, в 1 мл этого раствора содержится 20 мкг бензола. Из этого раствора готовят шкалу стандартов (табл. 37).
Таблица 37
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
|
Стандартный бутаноновый раствор, мл |
0 |
0,05 |
0,1 |
0,2 |
0,3 |
0,4 |
0,5 |
|
|
Бутанон, мл |
4 |
3,9 |
3,8 |
3,6 |
3,4 |
3,2 |
3,0 |
|
|
Содержание бензола, мкг |
0 |
1 |
2 |
4 |
6 |
8 |
10 |
Все пробирки шкалы обрабатывают аналогично пробе. Шкалой стандартов можно пользоваться для визуального определения, ее готовят в колориметрических пробирках одновременно с пробой. В первой пробирке может содержаться 0,5 мкг и далее, как указано в табл. 37.
Расчет см. выше.
ТОЛУОЛ
Бесцветная жидкость с характерным запахом. Плотность 0,867 при 20 °C, температура кипения 110,6 °C, смесь паров толуола с воздухом взрывоопасна. Толуол растворим в ряде органических растворителей.
Толуол обладает наркотическим действием, действует на нервную систему, кроветворные органы и др.
Предельно допустимые концентрации: максимальная разовая и среднесуточная 0,6 мг/м3.
ОПРЕДЕЛЕНИЕ ТОЛУОЛА НИТРОВАНИЕМ [2]
Принцип определения
При нитровании толуола образуется тринитротолуол, который в щелочной среде в присутствии кетона дает соединение, окрашенное в оранжево-розовый цвет.
Чувствительность определения — 2 мкг в определяемом объеме раствора. Другие ароматические углеводороды мешают определению.
Реактивы
1. Стандартный раствор толуола готовят из перегнанного. Исходный раствор получают в мерной колбе емкостью 50 мл, в которую внесено 1 — 2 мл нитрационной смеси. Колбу взвешивают, вносят 2 — 3 капли толуола и снова взвешивают. По разности двух взвешиваний устанавливают количество толуола. Раствор встряхивают и колбу помещают на 30 мин. в кипящую водяную баню. Раствор охлаждают и в него осторожно вливают воду. Колба в это время должна быть помещена в холодную водяную баню. Высчитывают содержание толуола в 1 мл раствора.
Рабочий стандартный раствор с содержанием 100 мкг/мл готовят соответствующим разведением исходного раствора. Объем доводят до метки нитрационной смесью, разбавленной водой в отношении 1:3. Другие реактивы и отбор пробы см. «Определение бензола нитрованием» выше.
Ход анализа
Поглотительный прибор с пробой помещают в кипящую водяную баню на 30 мин. По охлаждении раствор переливают в коническую колбу емкостью 50 мл, а поглотительный прибор ополаскивают несколько раз водой (5 — 6 мл).
Воду приливают к пробе осторожно, избегая сильного разогревания раствора.
После охлаждения пробу нейтрализуют 25% раствором аммиака. Нейтрализацию проводят из бюретки с краном при охлаждении в присутствии лакмусовой бумаги.
Затем пробу переводят в делительную воронку на 25 — 30 мин. с хорошо притертой пробкой, добавляют 2 мл бутанона и раствор встряхивают в течение 5 мин., дают расслоиться и водный нижний слой сливают через кран воронки, а верхний бутаноновый — через верх воронки в пробирку. Объем бутанонового раствора доводят до 2 мл бутаноном. В пробирку наливают 0,75 мл 20% раствора щелочи и содержимое пробирки встряхивают в течение нескольких минут. Через 15 — 20 мин. измеряют оптическую плотность раствора в кювете с толщиной слоя 0,5 — 1 см по сравнению с контролем, который готовят одновременно и аналогично пробе. Определение проводят с помощью фотоэлектроколориметра при длине волны 453 нм.
Содержание толуола в анализируемом объеме определяют по предварительно построенному калибровочному графику. Для построения графика готовят шкалу стандартов из стандартного бутанонового раствора, для получения его берут 1 мл рабочего стандартного раствора, выливают в коническую колбу емкостью 100 мл, в которую предварительно вливают 8 — 10 мл воды. Раствор охлаждают и нейтрализуют 25% раствором аммиака в присутствии лакмуса. Охлажденный нейтральный раствор выливают в делительную воронку и в нее вливают 10 мл бутанона. Раствор встряхивают в течение 10 мин. и оставляют в покое до полного расслоения раствора. Нижний водный слой сливают через кран воронки, а верхний бутаноновый слой переливают через верхний край воронки в мерную колбу емкостью 10 мл. Объем раствора доводят до метки бутаноном. 1 мл этого раствора содержит 10 мкг толуола. Из этого раствора готовят шкалу стандартов (табл. 38).
Таблица 38
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
|
Стандартный бутаноновый раствор, мл |
0 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
|
Тутанон, мл |
2 |
1,9 |
1,8 |
1,6 |
1,4 |
1,2 |
1,0 |
|
Содержание толуола, мкг |
0 |
1 |
2 |
4 |
6 |
8 |
10 |
Все пробирки шкалы обрабатывают аналогично пробе. Шкалой можно пользоваться для визуального определения, ее готовят в колориметрических пробирках одновременно с пробой.
Расчет см. выше.
КСИЛОЛ
|
C6H4(CH3)2 |
Мол. вес 106,17 |
Бесцветная жидкость. Ксилол может быть в виде трех изомеров: ортоксилол — температура кипения 144,4 °C, плотность при 20 °C 0,881; метаксилол — температура кипения 139,1 °C, плотность при 20° 0,864; параксилол, температура кипения 138,35 °C, плотность при 20 °C 0,861.
Пары ксилола тяжелее воздуха в 3,7 раза, взрывоопасны. Технический ксилол является смесью трех изомеров. Растворимость в воде при 22 °C 0,013%.
Ксилол — наркотик, действие его на организм человека сходно с бензолом и толуолом, действуют они на кроветворные органы.
Предельно допустимая концентрация: максимальная разовая и среднесуточная 0,2 мг/м3.
ОПРЕДЕЛЕНИЕ КСИЛОЛА НИТРОВАНИЕМ [3]
Принцип определения
При нитровании ксилола образуется нитросоединение ксилола, которое в щелочной среде в присутствии кетона дает соединение, окрашенное в серо-зеленый цвет.
Чувствительность определения 2 мкг в определяемом объеме раствора. Другие ароматические соединения мешают определению.
Реактивы
1. Стандартный раствор готовят из свежеперегнанного ксилола при 138 — 145 °C. Исходный раствор получают в мерной колбе емкостью 50 мл, в которой помещено 1 — 5 мл нитрационной смеси. Колбу взвешивают и в нее вносят 2 — 3 капли ксилола. По разности двух взвешиваний устанавливают количество ксилола в 1 мл. Колбу помещают в кипящую водяную баню на 30 мин. По охлаждении в колбу вносят малыми порциями дистиллированную воду до метки.
Рабочий стандартный раствор с содержанием 100 мкг/мл готовят в мерной колбе емкостью 50 мл соответствующим разбавлением исходного стандартного раствора.
Раствор доводят до метки нитрационной смесью, разбавленной водой в отношении 1:3.
Другие реактивы и отбор пробы см. «Определение бензола нитрованием» выше.
Ход анализа
Поглотительный прибор с пробой помещают в кипящую водяную баню на 30 мин. По охлаждении раствора его переливают в коническую колбу емкостью 50 мл и поглотительный прибор ополаскивают несколько раз водой, на что расходуют 5 — 6 мл. Воду приливают к пробе осторожно, избегая сильного разогревания.
После охлаждения пробу нейтрализуют 25% раствором аммиака. Нейтрализацию проводят из бюретки с краном при охлаждении раствора в присутствии лакмуса. Затем пробу переводят в делительную воронку емкостью 25 — 30 мл с хорошо притертой пробкой, добавляют к пробе 2,2 мл бутанона и раствор встряхивают в течение 5 мин. Раствору дают расслоиться и водный нижний слой сливают через кран воронки, а верхний, бутаноновый, переливают через верх воронки в пробирку. Объем бутанонового раствора доводят до 2 мл бутаноном. В пробирку наливают 0,75 мл щелочи и содержимое пробирки встряхивают в течение нескольких минут. Через 15 — 20 мин. измеряют оптическую плотность в кювете толщиной слоя 0,5 — 1 см по сравнению с контролем, который готовят одновременно и аналогично. Определение проводят с помощью фотоэлектроколориметра при длине волны 508 нм.
Содержание ксилола в анализируемом объеме определяют по предварительно построенному калибровочному графику. Для построения графика готовят шкалу стандартов из стандартного бутанонового раствора, для получения его берут 1 мл рабочего стандартного раствора, выливают в коническую колбу емкостью 100 мл, в которую было влито 8 — 10 мл воды. Раствор охлаждают и нейтрализуют 25% раствором аммиака в присутствии лакмуса.
Охлажденный нейтральный раствор выливают в делительную воронку и в нее вливают 10 мл бутанона. Растворы встряхивают в течение 10 мин. и оставляют в покое до полного разделения слоев. Нижний водяной слой сливают через кран, а верхний бутаноновый через верх воронки сливают в мерную колбу емкостью 10 мл или в пробирку с притертой пробкой и меткой 10 мл. Раствор доводят до метки бутаноном, в 1 мл этого раствора содержится 10 мкг ксилола.
Из этого раствора готовят шкалу стандартов (табл. 39).
Таблица 39
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
|
Стандартный бутаноновый раствор, мл |
0 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
|
|
Бутанон, мл |
2 |
1,8 |
1,6 |
1,4 |
1,2 |
1,0 |
|
|
Содержание ксилола, мкг |
0 |
2 |
4 |
6 |
8 |
10 |
Все пробирки шкалы обрабатывают аналогично пробе.
Шкалой стандартов можно пользоваться для визуального определения, ее готовят в колориметрических пробирках одновременно с пробой.
Расчет см. выше.
ИЗОПРОПИЛБЕНЗОЛ
|
C6H5CH(CH3)2 |
Мол. вес 120,19 |
Бесцветная жидкость, температура кипения 152,39 °C, плотность 0,86 при 20 °C. Хорошо растворим в органических растворителях. В воде не растворим.
Изопропилбензол токсичен.
Предельно допустимые концентрации: максимальная разовая и среднесуточная 0,014 мг/м3.
ОПРЕДЕЛЕНИЕ ИЗОПРОПИЛБЕНЗОЛА НИТРОВАНИЕМ [4]
Принцип определения
При взаимодействии изопропилбензола с нитрационной смесью образуется полинитросоединение. После извлечения нитросоединения эфиром, растворения сухого остатка в этаноле в присутствии щелочи раствор окрашивается в оранжевый цвет.
Чувствительность определения — 1 мкг изопропилбензола в определяемом объеме раствора. Другие ароматические соединения мешают определению.
Реактивы
1. Стандартный раствор готовят из свежеперегнанного изопропилбензола. Исходный стандартный раствор готовят в мерной колбе емкостью 25 — 50 мл, в которую вносят 5 — 10 мл нитрационной смеси. Колбу взвешивают, вносят 1 — 2 капли изопропилбензола и вновь взвешивают.
По разности взвешиваний устанавливают количество изопропилбензола. Раствор встряхивают и помещают в кипящую водяную баню на 1 ч. После охлаждения раствора его доводят до метки нитрационной смесью. Рассчитывают содержание изопропилбензола в 1 мл.
Рабочий стандартный раствор с содержанием 100 мкг/мл готовят соответствующим разбавлением исходного нитрационной смесью.
Приготовление других реактивов и отбор пробы см. «Определение бензола нитрованием» выше.
Для установления содержания изопропилбензола в пределах предельно допустимой концентрации необходимо протянуть не менее 70 л исследуемого воздуха.
Ход анализа
а) Извлечение нитросоединения бутаноном
Поглотительный прибор с пробой помещают в кипящую водяную баню на 1 ч. По охлаждении раствора его переливают в коническую колбу емкостью 50 мл и поглотительный прибор ополаскивают несколько раз водой, на что расходуют 5 — 6 мл, воду приливают к пробе осторожно, избегая сильного разогревания раствора.
После охлаждения раствора пробу нейтрализуют 25% раствором аммиака. Нейтрализацию проводят из бюретки с краном при охлаждении в присутствии лакмуса. Затем пробу переводят в делительную воронку, добавляют 2,2 мл бутанона и раствор встряхивают в течение 5 мин. Жидкость оставляют в покое для расслоения. Нижний водный слой сливают через кран воронки, а верхний, бутаноновый, переливают в пробирку через верх воронки. Объем бутанонового раствора доводят до 2 мл бутаноном. В пробирку наливают 0,75 мл щелочи, содержимое пробирки встряхивают в течение нескольких минут и через 15 — 30 мин. сравнивают интенсивность окраски пробы со шкалой стандартов, которую готовят одновременно с пробой.
Шкалу стандартов готовят из стандартного бутанонового раствора, для получения его берут 1 мл рабочего стандартного раствора, выливают в коническую колбу емкостью 100 мл, в которую предварительно влито 8 — 10 мл воды. Раствор охлаждают и нейтрализуют 25% раствором аммиака в присутствии лакмуса.
Охлажденный нейтральный раствор выливают в делительную воронку, в нее вливают 10 мл бутанона. Раствор встряхивают в течение 10 мин. и оставляют в покое до полного разделения слоев. Нижний, водный, раствор сливают через кран воронки, а верхний, бутаноновый, — в мерную колбу емкостью 10 мл или в пробирку с притертой пробкой и меткой 10 мл. Объем раствора доводят до метки бутаноном. Получают стандартный бутаноновый раствор с содержанием 10 мкг/мл, из него готовят шкалу стандартов (табл. 40). Все пробирки шкалы обрабатывают аналогично и одновременно с пробой.
Таблица 40
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
|
Стандартный бутаноновый раствор, мл |
0 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
|
|
Бутанон, мл |
2 |
1,9 |
1,8 |
1,6 |
1,4 |
1,2 |
1,0 |
|
|
Содержание изопропилбензола, мкг |
0 |
1 |
2 |
4 |
6 |
8 |
10 |
б) Извлечение нитросоединения эфиром
Поглотительный прибор с пробой помещают на 1 ч в кипящую водяную баню, затем охлаждают и переводят в коническую колбу емкостью 50 мл и поглотительный прибор ополаскивают водой несколько раз, на что расходуют 5 — 6 мл воды. Промывную воду приливают к пробе осторожно, избегая сильного разогревания раствора.
По охлаждении содержимое колбы переливают в делительную воронку и добавляют 5 мл эфира. Раствор встряхивают в течение 5 мин. и оставляют в покое для полного разделения слоев. Затем водный, нижний, слой сливают через кран воронки, а верхний оставляют в воронке и к нему приливают 5 мл воды. Раствор вновь встряхивают. После разделения растворов нижний слой, водный, выливают через кран воронки, а верхний, эфирный, переливают через верх воронки в фарфоровую чашку и испаряют эфир.
Сухой остаток постепенно растворяют в 2 мл этанола и раствор переносят в пробирку с меткой 2 мл. В пробирку вливают 0,5 мл 5% раствора спиртовой щелочи и встряхивают в течение нескольких минут.
Одновременно готовят шкалу стандартов (табл. 41).
Таблица 41
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
|
Рабочий стандартный раствор, мл |
0 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
0,9 |
|
Этанол, мл |
2 |
1,9 |
1,8 |
1,6 |
1,6 |
1,2 |
1,0 |
|
Содержание изопропилбензола, мкг |
0 |
1 |
2 |
4 |
6 |
8 |
10 |
Все пробирки шкалы обрабатывают аналогично пробе.
При извлечении нитросоединения, как эфиром, так и бутаноном, определение проводят визуально через 25 — 30 мин. после добавления щелочи.
Расчет см. выше.
ЭТИЛБЕНЗОЛ
Прозрачная бесцветная жидкость. Плотность при 20 °C 0,86, температура кипения 136,15 °C. Этилбензол плохо растворим в воде, в 100 мл воды при 15 °C растворяется 4,014 г, в спирте и эфире растворим хорошо. Пары этилбензола вызывают слезотечение и боль в глазах.
Предельно допустимые концентрации: максимальная разовая и среднесуточная 0,02 мг/м3.
ОПРЕДЕЛЕНИЕ ЭТИЛБЕНЗОЛА НИТРОВАНИЕМ [6]
Принцип определения
При взаимодействии этилбензола с нитрационной смесью образуется тринитроэтилбензол, который в щелочной среде в присутствии кетона дает окрашенное соединение.
Чувствительность определения 1 мкг этилбензола в анализируемом объеме раствора. Другие ароматические соединения мешают определению.
Реактивы
1. Стандартный исходный раствор этилбензола.
В мерную колбу емкостью 25 мл вносят 10 мл нитрационной смеси. Колбу взвешивают, в нее вливают 1 — 2 капли этилбензола и снова взвешивают. По разности двух взвешиваний определяют количество этилбензола в колбе. Колбу помещают на 30 мин. в кипящую водяную баню и по охлаждении раствора доводят его объем до метки нитрационной смесью и хорошо перемешивают.
Высчитывают содержание этилбензола в 1 мл раствора.
Рабочий стандартный раствор с содержанием 10 мкг/мл готовят в мерной колбе емкостью 25 мл из исходного стандартного раствора. Для этого берут такое количество этого раствора, в котором содержится 250 мкг этилбензола. Объем жидкости в колбе доводят до метки нитрационной смесью.
Другие реактивы и отбор пробы см. «Определение бензола нитрованием» выше.
Для определения этилбензола в пределах максимальной разовой концентрации необходимо протянуть не менее 50 мл исследуемого воздуха.
Ход анализа
Поглотительный прибор с пробой помещают на 30 мин. в кипящую водяную баню. По истечении этого времени пробу охлаждают и осторожно переливают в делительную воронку, в которую предварительно наливают 6 — 8 мл воды. При вливании необходимо избегать сильного разогревания раствора. Поглотительный прибор промывают дважды по 2 мл воды. Воду каждый раз осторожно вливают в делительную воронку.
По охлаждении раствора в воронке в нее вливают 5 мл эфира. Раствор встряхивают в течение 5 мин. и оставляют в покое до полного расслоения. Нижний, водный, слой сливают через кран, верхний оставляют в воронке, к нему приливают 5 мл воды, насыщенной эфиром, и взбалтывают для удаления из эфирного слоя следов серной кислоты. После расслоения растворы разделяют — эфирный выливают в чашку и испаряют эфир почти досуха при комнатной температуре в вытяжном шкафу вдали от огня.
Остаток эфирного раствора переводят в колориметрическую пробирку с притертой пробкой и объем раствора доводят эфиром до 1 мл; предварительно этим эфиром ополаскивают фарфоровую чашку. Затем фарфоровую чашку ополаскивают 1 мл этанола и раствор выливают в ту же пробирку. Объем доводят до 2 мл. Содержимое пробирки перемешивают, вливают 0,1 мл 5% спиртового раствора щелочи и вновь встряхивают. Через 10 — 15 мин. интенсивность окраски пробы сравнивают с окраской шкалы стандартов, которую готовят одновременно с пробой (табл. 42).
Таблица 42
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
|
Рабочий стандартный раствор, мл |
0 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
|
Нитросмесь, мл |
1 |
0,9 |
0,8 |
0,6 |
0,4 |
0,2 |
0 |
|
Содержание этилбензола, мкг |
0 |
1 |
2 |
4 |
6 |
8 |
10 |
Каждую пробирку шкалы обрабатывают аналогично пробе.
Расчет см. выше.
НИТРОБЕНЗОЛ
Бесцветная жидкость с запахом горького миндаля, температура кипения 210,9 °C, температура плавления 5,7 °C. Нитробензол слабо растворим в воде, хорошо растворяется в спирте, эфире; перегоняется с водяным паром.
Нитробензол токсичен, отравление возможно при вдыхании паров и через кожу; он является кровяным ядом.
Предельно допустимые концентрации: максимальная разовая и среднесуточная 0,008 мг/м3.
ОПРЕДЕЛЕНИЕ НИТРОБЕНЗОЛА НИТРОВАНИЕМ [5]
Принцип определения
При взаимодействии нитробензола с нитрационной смесью образуется динитросоединение, которое в щелочной среде в присутствии кетона дает соединение, окрашенное в красно-фиолетовый цвет.
Чувствительность определения — 1 мкг в анализируемом объеме раствора. Ароматические соединения мешают определению.
Реактивы
1. Стандартный раствор нитробензола: готовят исходный раствор в мерной колбе емкостью 50 мл, в которую влито 12 мл нитрационной смеси. Колбу взвешивают и вносят в нее 2 — 3 капли нитробензола и вновь взвешивают. По разности двух взвешиваний устанавливают количество нитробензола в колбе. Раствор встряхивают и помещают в кипящую водяную баню на 40 мин. По истечении этого времени раствор охлаждают и постепенно, осторожно в колбу вливают воду при помешивании раствора. Колбу в это время помещают в холодную водяную баню. Раствор доводят до метки после того, как температура его сравняется с комнатной. Высчитывают содержание нитробензола в 1 мл раствора.
Рабочий стандартный раствор с содержанием 100 мкг/мл готовят в мерной колбе емкостью 50 мл, в нее вливают такое количество исходного раствора, которое содержит 5000 мкг нитробензола. Раствор доводят до метки нитрационной смесью, разбавленной водой в отношении 1:3.
2. Нитробензол свежеперегнанный, температура кипения 210,9 °C.
Приготовление других реактивов и отбор пробы см. «Определение бензола нитрованием» выше. Для определения нитробензола в пределах предельно допустимой концентрации необходимо протянуть не менее 70 л исследуемого воздуха.
Ход анализа
Поглотительный прибор с пробой помещают в кипящую водяную баню на 40 мин. По охлаждении раствора его сливают в коническую колбу емкостью 50 мл и поглотительный прибор ополаскивают несколько раз водой, на что расходуют 6 — 8 мл. Воду приливают к пробе осторожно, избегая сильного разогревания.
После охлаждения раствора его нейтрализуют 25% раствором аммиака по лакмусу. Нейтрализацию проводят из бюретки с краном при охлаждении раствора. Затем пробу переводят, в делительную воронку емкостью 25 — 30 мл и добавляют 2,2 мл бутанона. Раствор встряхивают в течение 5 — 10 мин. и дают расслоиться. Водный нижний слой сливают через кран воронки, верхний бутаноновый слой переливают через верхний край воронки в пробирку и объем раствора доводят до 2 мл бутаноном.
В раствор вливают 0,75 мл 20% раствора щелочи, содержимое пробирки встряхивают в течение нескольких минут и через 15 — 20 мин. измеряют оптическую плотность в кювете с толщиной слоя 0,5 см по сравнению с контролем, который готовят одновременно и аналогично пробе. Определение проводят при длине волны 536 нм.
Содержание нитробензола в анализируемом объеме определяют по предварительно поставленному калибровочному графику. Для построения графика готовят шкалу стандартов из стандартного бутанонового раствора, для получения его берут 1 мл рабочего стандартного раствора, вливают осторожно в коническую колбу емкостью 100 мл, в которую предварительно влито 8 — 10 мл воды. Раствор охлаждают и нейтрализуют 25% раствором аммиака в присутствии лакмуса.
Охлажденный нейтральный раствор переливают в делительную воронку, к нему приливают 10 мл бутанона. Раствор встряхивают в течение 10 мин. и оставляют в покое до полного разделения слоев. Затем нижний водный слой удаляют через кран воронки, а верхний бутаноновый — через верх воронки, вливают в мерную колбу емкостью 10 мл или в пробирку с притертой пробкой и меткой 10 мл. Этот раствор содержит нитробензола 10 мкг/мл.
Из этого раствора готовят шкалу стандартов (табл. 43).
Таблица 43
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
|
Стандартный бутаноновый раствор, мл |
0 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
|
Бутанон, мл |
2 |
1,9 |
1,8 |
1,6 |
1,4 |
1,2 |
1,0 |
|
Содержание нитробензола, мкг |
0 |
1 |
2 |
4 |
6 |
8 |
10 |
Все пробирки шкалы обрабатывают аналогично пробе.
Шкалой стандартов можно пользоваться для визуального определения, ее готовят в колориметрических пробирках одновременно с пробой.
Расчет см. выше.
ДИНИЛ
Динил — это смесь 26,5% раствора дифенила и 73,5% раствора дифинилового эфира. Жидкость с характерным резким запахом, температура кипения 258 °C.
Динил является токсичным веществом, он вызывает изменение в почках, печени и в других органах.
Предельно допустимые концентрации: максимальная разовая и среднесуточная 0,01 мг/м3.
ОПРЕДЕЛЕНИЕ ДИНИЛА НИТРОВАНИЕМ [8]
Принцип определения
Метод основан на определении окрашенного продукта реакции, образовавшегося при взаимодействии нитропроизводного динила с ацетоном и сульфидом натрия.
Чувствительность определения — 0,25 мкг в анализируемом объеме раствора. Метадинитробензол определению не мешает. Влияние других гомологов бензола устраняют применением хромовой смеси для их окисления в процессе анализа.
Реактивы
1. Поглотительный раствор — нитрационная смесь. 10 г нитрата калия и 0,5 г сернистокислого натрия растворяют в 100 мл серной кислоты уд. веса 1,84.
2. Стандартный раствор динила. Исходный раствор готовят в мерной колбе емкостью 50 мл, в которую наливают 1 — 2 мл нитрационной смеси, колбу взвешивают, вносят 2 — 3 капли динила и снова взвешивают. По разности устанавливают количество динила в колбе и рассчитывают его содержание в 1 мл раствора. Стандартный раствор с содержанием 100 мкг/мл и рабочий стандартный раствор с содержанием 10 мг/мл готовят соответствующим разведением нитросмесью.
3. Толуол.
4. Ацетон.
5. Натрий углекислый, 2% раствор, подкрашенный фенолфталеином до розовой окраски.
6. Натрий сернистый, 1% раствор водный или спиртовой (хранят в закрытой склянке не более месяца).
7. Уксусная кислота, 10% раствор.
8. Калий двухромовокислый, 10% раствор.
9. Фенолфталеин, 0,1% раствор.
Отбор пробы
а) Для определения максимальной разовой концентрации воздух со скоростью 1 л/мин. протягивают через поглотительный прибор с пористым фильтром, наполненный 2,5 мл нитрационной смеси. Продолжительность отбора 20 — 30 мин.
б) Для определения среднесуточной концентрации воздух со скоростью 0,5 л/мин. протягивают через поглотительный прибор с 5 мл нитрационной смеси в течение суток. Допустимо проводить отбор в один и тот же поглотительный прибор с перерывами в 2 — 4 ч.
Ход анализа
В поглотительный прибор вводят 0,5 мл воды, перемешивают, используя для этого резиновую грушу. Прибавляют 1 мл 10% раствора двухромовокислого калия, перемешивают и помещают на 30 мин. в кипящую водяную баню. При отборе пробы в 5 мл нитрационной смеси в поглотитель вводят 1 мл воды и 2 мл раствора бихромата.
После нагрева в поглотитель вводят 10 мл воды, охлаждают и сливают в делительную воронку. Поглотительный прибор ополаскивают 2 раза по 2 мл воды, сливая ее в воронку. В воронку вводят 2 мл толуола для экстрагирования и интенсивно взбалтывают в течение 2 — 3 мин. После расслоения водный слой отделяют, а экстракт промывают 3 — 5 мл раствора углекислого натрия. При обесцвечивании раствора соды его прибавляют в количестве 1 — 2 мл.
Содовый слой отделяют, экстракт сливают через верх воронки в пробирку. Делительную воронку ополаскивают 0,5 мл толуола, который присоединяют к экстракту. В пробирку добавляют ацетона до 10 мл и 0,05 мл 1% раствора сульфида натрия, перемешивают и через 2 мин. подкисляют 0,3 мл 10% раствора уксусной кислоты. Через 5 — 10 мин. фотометрируют в кювете с толщиной слоя 1 см при длине волны 450 нм по сравнению с контролем. Контроль готовят одновременно и аналогично пробе. Содержание динила определяют по заранее построенному калибровочному графику. Для построения калибровочного графика готовят шкалу стандартов (табл. 44).
Таблица 44
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
|
Рабочий стандартный раствор, мл |
0 |
0,025 |
0,05 |
0,075 |
0,1 |
0,15 |
0,2 |
|
Нитрационная смесь, мл |
2,5 |
2,475 |
2,450 |
2,425 |
2,4 |
2,35 |
2,25 |
|
Количество динила, мкг |
0 |
0,25 |
0,5 |
0,75 |
1,0 |
1,5 |
2,0 |
Все пробирки шкалы обрабатывают аналогично пробам, измеряют оптическую плотность и строят график.
Расчет см. выше.
СТИРОЛ
|
C6H5·CH=CH2 |
Мол. вес 104,15 |
Бесцветная жидкость с температурой кипения 146 °C, температурой плавления 33 °C. Плотность ее 0,909 (20°); упругость пара 10 мм рт. ст. (33°). Стирол трудно растворим в воде, хорошо растворим в спирте, эфире, метаноле, ацетоне и других органических растворителях. Стирол полимеризуется в полистирол, особенно этот процесс ускоряется с повышением температуры. Стирол обладает наркотическим действием, раздражающим действием на слизистые оболочки, вызывает поражение печени, а также действует на кроветворные органы. Предельно допустимые концентрации стирола: максимальная разовая 0,003 мг/м3, среднесуточная 0,003 мг/м3.
ОПРЕДЕЛЕНИЕ СТИРОЛА ХРОМАТОГРАФИЕЙ НА БУМАГЕ [7]
Принцип определения
При взаимодействии стирола с ацетатом ртути образуется ртутно-органическое производное, которое выделяют из смеси на бумаге нисходящим способом в системе растворителей н-бутанол — вода — диэтиламин. При орошении хроматограммы раствором дифенилкарбазида производные стирола проявляются в виде фиолетовых зон, Rf = 0,8.
Количественное определение стирола проводят фотометрически по измерению оптической плотности элюата или визуально, сравнивая интенсивность окраски зон локализации пробы и шкалы.
Чувствительность определения 1 мкг стирола в анализируемом объеме раствора. Изопропилбензол, бензол, гидроперекись изопропилбензола, перекись бензоила, малеиновый, фталевый ангидриды, диметиланилин, дивинил, аммиак, дитолилметан, изопрен, хлоропрен не мешают определению.
Аппаратура
1. Теплоэлектровентилятор.
2. Хроматографическая камера, размер 240 на 600 мм.
3. Микропипетки емкостью 0,1 — 0,2 мл с делениями на 0,001 мл.
4. Пробирки высотой 9 см, диаметром 0,8 см с делениями 0,1 — 0,2 — 0,3 мл.
5. Стеклянный распылитель.
Реактивы
1. Поглотительный раствор. К 100 мл этанола добавляют 0,1 г ацетата ртути и 0,5 мл уксусной кислоты.
2. Ацетат ртути, двузамещенная соль.
3. Этиловый спирт, очищенный. К 1 л этанола добавляют 40 г едкого кали, оставляют на 2 ч, перегоняют при 78 °C.
4. Кали едкое, х.ч.
5. н-Бутанол.
6. Диэтиламин.
7. Дифенилкарбазид, 0,1% раствор в этаноле.
8. Хроматографическая бумага «Медленная».
9. Стандартный раствор стирола. Исходный раствор готовят из свежеперегнанного стирола (при 145 °C). В мерную колбу вносят 2 — 3 мл поглотительного раствора, взвешивают. Добавляют 1 — 2 капли стирола и снова взвешивают. Объем 25 мл колбы доводят до метки поглотительным раствором. Рассчитывают содержание стирола в 1 мл. Соответствующим разбавлением поглотительным раствором готовят раствор N 1 с содержанием 100 мкг/мл.
10. Система растворителей. Готовят смесь н-бутанола, воды, диэтиламина, 5:4:1. Смесь помещают в делительную воронку, перемешивают. После разделения слоев нижний используют для смачивания фильтровальной бумаги с целью насыщения хроматографической камеры парами растворителей. Верхний слой используют для наполнения лодочки.
Отбор пробы
Воздух со скоростью 6 л/мин. протягивают через три пары параллельно соединенных поглотительных приборов Зайцева, содержащих по 2 мл поглотительного раствора. Отбирают не менее 60 л исследуемого воздуха при охлаждении. По мере улетучивания спирта в процессе отбора в поглотители периодически вносят спирт, подкисленный уксусной кислотой, до первоначального объема.
Ход анализа
Жидкость из первых трех поглотительных приборов переносят в выпаривательные чашки, смывают поглотительные приборы по 1 мл подкисленным спиртом, промывные жидкости объединяют с пробой. Аналогично поступают со вторыми поглотительными приборами.
Удаляют спирт выпариванием при комнатной температуре или при нагревании не выше 50 °C, так, чтобы осадок оставался влажным. Остаток в чашке количественно переносят в пробирку, обмывают чашку спиртом, подкисленным уксусной кислотой, и доводят объем до 0,3 мл.
В хроматографической камере по стенкам располагают фильтровальную бумагу, смоченную нижним слоем жидкости, полученной при приготовлении системы растворителей.
Верхний слой жидкости помещают в лодочку, которая расположена в хроматографической камере. Насыщение хроматографической камеры проходит в течение 3 — 4 ч. На лист хроматографической бумаги на расстоянии 7 см от нижнего края намечают линию старта, на которой фиксируют точки нанесения пробы, на расстоянии 3 — 3,5 см друг от друга.
С помощью микропипетки в указанные точки наносят пробу. В одну точку наносят содержимое первых поглотительных приборов, а во вторую — содержимое вторых, контрольных, поглотителей.
Наносят по 0,15 или 0,3 мл пробы в зависимости от содержания стирола. Если наносят всю пробу, то чашку обмывают этиловым спиртом. При нанесении растворов на бумагу ее обдувают теплым воздухом при помощи вентилятора.
Хроматографическую бумагу с пробой помещают в лодочку. Камеру герметизируют и оставляют при комнатной температуре на 14 — 15 ч. По истечении времени растворитель опускается на расстояние 25 см. Бумагу извлекают из камеры, высушивают на воздухе (0,5 — 1 ч) и орошают раствором дифенилкарбазида. Хроматограмму помещают в термостат и выдерживают в течение 1 — 2 мин. при 70 °C. При наличии стирола на хроматограмме проявляются окрашенные в фиолетовый цвет зоны локализации в виде круглых пятен (Rf = 0,8).
На линии старта также проявляются окрашенные зоны, свидетельствующие об избытке ацетата ртути. Зоны, характерные для стирола, вырезают из бумаги, измельчают и помещают в пробирки, в которые вносят по 3,5 мл очищенного этанола. Содержимое пробирок встряхивают, через 5 и 15 мин. элюаты фотометрируют на спектрофотометре в кюветах с толщиной слоя 1 см при длине волны 570 нм. Содержание стирола устанавливают по калибровочному графику.
Для построения калибровочного графика готовят шкалу стандартов (табл. 45).
Таблица 45
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
|
Стандартный раствор N 1, мл |
0 |
0,33 |
1,0 |
1,66 |
3,33 |
5 |
|
Поглотительный раствор, мл |
5 |
4,67 |
4,0 |
3,34 |
1,67 |
0 |
|
Содержание стирола в объеме 0,15 мл, мкг |
0 |
1,0 |
3,0 |
5,0 |
10,0 |
15,0 |
Согласно шкале стандартов (см. табл. 45), получают ряд растворов 1 — 2 — 3…5 с содержанием от 1 до 15 мкг стирола в объеме 0,15 мл.
На хроматографическую бумагу по линии старта наносят по 0,15 мл каждого раствора, подвергают хроматографированию и проводят все операции аналогично пробе. На основании данных оптических плотностей строят калибровочный график.
Шкалой стандартов можно пользоваться при визуальном определении.
Расчет см. выше.
![]() -МЕТИЛСТИРОЛ
-МЕТИЛСТИРОЛ
|
|
Мол. вес 118,18 |
Бесцветная или слегка желтоватая жидкость с неприятным запахом. Температура кипения 162 — 163 °C, удельный вес 0,914, в воде не растворим, растворим в органических растворителях.
Пары альфа-метилстирола вызывают раздражение глаз и верхних дыхательных путей. Жидкий ![]() -метилстирол вызывает раздражение кожных покровов.
-метилстирол вызывает раздражение кожных покровов.
Предельно допустимые концентрации: максимальная разовая и среднесуточная 0,04 мг/м3.
ОПРЕДЕЛЕНИЕ ![]() -МЕТИЛСТИРОЛА ХРОМАТОГРАФИЕЙ НА БУМАГЕ
-МЕТИЛСТИРОЛА ХРОМАТОГРАФИЕЙ НА БУМАГЕ
![]() -Метилстирол определяют аналогичным стиролу методом (см. выше).
-Метилстирол определяют аналогичным стиролу методом (см. выше).
ГИДРОПЕРЕКИСЬ ИЗОПРОПИЛБЕНЗОЛА
|
C6H5(CH3)2COOH |
Мол. вес 152,19 |
Жидкость бесцветная маслянистая, запах ее напоминает озон. Плотность 1,062 (20°), температура кипения 60 °C (0,2 мм), температура разложения 741°, воспламенения 80°. Взрывает при 170 °C. Растворима в органических растворителях, легко растворима в воде. Под действием кислот и катализаторов разлагается с образованием эквимолекулярных количеств фенола и ацетона.
Гидроперекись изопропилбензола вызывает раздражение слизистых оболочек, покраснение кожи, одышку, изменение состава крови.
Предельно допустимые концентрации: максимальная разовая и среднесуточная 0,007 мг/м3.
ОПРЕДЕЛЕНИЕ ГИДРОПЕРЕКИСИ ИЗОПРОПИЛБЕНЗОЛА ПО ФЕНОЛУ [9]
Принцип определения
При действии серной кислоты на гидроперекись изопропилбензола образуется фенол, который взаимодействует с П-нитрофенилдиазонием и дает продукт, окрашенный в красный цвет. Чувствительность определения 1,5 мкг в определяемом объеме раствора. Мешают определению фенол, крезолы, амины.
Реактивы
1. Силикагель, КСМ, МСМ, КСК, МСК, размер зерен 0,25 — 0,5 мм.
2. Этанол, предварительно обработанный (см. выше).
3. Стандартный раствор гидроперекиси изопропилбензола (ГПИПБ). Исходный стандартный раствор готовят из 99,9% ГПИПБ в мерной колбе емкостью 25 мл.
В колбу вносят 5 мл этанола, взвешивают, затем добавляют 1 — 3 капли ГПИПБ и вновь взвешивают. Объем колбы доводят до метки этанолом. Расчетным путем устанавливают содержание его в 1 мл. Исходный раствор устойчив в течение месяца. Рабочий стандартный раствор с содержанием 10 мкг/мл готовят в мерной колбе емкостью 100 мл. Для этого в колбу вносят расчетное количество исходного стандартного раствора 0,5 мл этанола и 2 мл серной кислоты уд. веса 1,84. Колбу нагревают на водяной бане при температуре 70° в течение часа, при этом колба должна находиться в наклонном положении и прикрыта обратной стороной притертой пробки. По отстаивании объем колбы доводят до метки водой. Раствор употребляют свежеприготовленный.
4. Соляная кислота, уд. вес 1,19.
5. Серная кислота, уд. вес 1,84.
6. Карбонат натрия насыщенный, 0,8% раствор.
7. Нитрит натрия, 25% раствор.
8. Паранитроанилин.
9. Паранитрофенилдиазоний. Готовят добавлением к 50 мл воды 2,5 мл соляной кислоты уд. веса 1,19, 2,5 мл 25% раствора нитрита натрия и 0,012 паранитроанилина. Тщательно перемешивают. Употреблять свежеприготовленный.
Отбор пробы
а) Для определения разовой концентрации отбор проб проводят в кипящий слой силикагеля.
Для этого по 3 мл силикагеля через воронку вносят в поглотительные приборы Яворовской или НИИ гигиены имени Ф.Ф. Эрисмана. Два последовательно присоединенных прибора в свою очередь соединяют с поглотителем, служащим для улавливания переброшенных зерен силикагеля. Воздух протягивают со скоростью до 10 л/мин. в течение 20 — 30 мин.
б) Для определения среднесуточной концентрации воздух протягивают со скоростью до 5 л/мин. через 2 последовательно соединенных поглотительных прибора, содержащих по 3 мл силикагеля. Отбор проб проводят непрерывно в течение суток.
Ход анализа
Силикагель из каждого поглотительного прибора отдельно переносят в пробирки с притертой пробкой.
Переброшенные зерна силикагеля присоединяют к содержимому второго поглотителя.
Добавляют по 4 мл этанола. Периодически в течение 30 мин. встряхивают содержимое пробирок. Для анализа берут по 2 мл прозрачного раствора и помещают их в пробирки с притертыми пробками, добавляют по 0,1 мл серной кислоты уд. веса 1,84 и нагревают пробирки при 70 °C в течение часа на водяной бане, поместив пробирки в наклонном положении и прикрыв их обратной стороной притертой пробки.
После охлаждения проводят нейтрализацию, добавляя по каплям насыщенный раствор карбоната натрия до щелочной реакции по лакмусу. Объем пробы доводят до 5 мл 0,8% раствором карбоната натрия. Добавляют 0,1 мл раствора n-нитрофенилдиазония. Содержимое перемешивают и через 10 мин. сравнивают окраску проб со шкалой стандартов (табл. 46), которую готовят одновременно и аналогично пробам.
Таблица 46
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
|
Рабочий стандартный раствор, мл |
0 |
0,15 |
0,3 |
0,4 |
0,6 |
0,8 |
1,0 |
1,5 |
|
Насыщенный раствор карбоната натрия, мл |
До нейтрализации (по лакмусу) |
|||||||
|
Раствор карбоната натрия, 0,8%, мл |
До объема 5 мл |
|||||||
|
Содержание ГПИПБ, мкг |
1,5 |
3,0 |
4,0 |
6,0 |
8,0 |
10,0 |
15,0 |
Расчет см. выше.
ЛИТЕРАТУРА
1. Алексеева М.В. Определение бензола в воздухе. Заводская лаборатория, 1942, 9.
2. Алексеева М.В. Определение толуола в воздухе. Заводская лаборатория, 1942, 9.
3. Определение ксилола. В сб.: Предельно допустимые концентрации атмосферных загрязнений. М., 1963, в. 7.
4. Алексеева М.В. Определение изопропилбензола. В кн.: Определение атмосферных загрязнений. М., Медгиз, 1963.
5. Алексеева М.В. Определение нитробензола. В сб.: Предельно допустимые концентрации атмосферных загрязнений. М., 1964, в. 8.
6. Гронсберг Е.Ш. Определение этилбензола. В сб.: Определение вредных веществ в воздухе производственных помещений. Горький, 1960.
7. Казнина Н.И. Определение стирола в воздухе с помощью бумажной хроматографии. Гиг. и сан., 1968, 5.
8. Марич Ю.А., Борский В.И. Метод определения динила в воздухе производственных помещений. Гиг. и сан., 1971, 1.
9. Хрусталева В.А. Определение гидроперекиси изопропилбензола в воздухе. В сб.: Ученые записки Московского научно-исследовательского института санитарии и гигиены имени Ф.Ф. Эрисмана, 1960, 5.
Глава IX
ХЛОРОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
Могут встречаться в виде газов, жидкостей и твердых веществ. Большая их часть — жидкости бесцветные, мало растворимы в воде, хорошо растворимы в спирте, эфире.
Хлорорганические соединения — углеводороды, являются наркотиками, некоторые действуют на внутренние органы (печень, почки), а также на нервную систему. Предельно допустимые концентрации некоторых хлорированных соединений даны в табл. 47.
Таблица 47
ПРЕДЕЛЬНО ДОПУСТИМЫЕ КОНЦЕНТРАЦИИ
|
Наименование |
Максимальная разовая, мг/м3 |
Среднесуточная, мг/м3 |
|
Дихлорэтан |
3 |
1 |
|
Трихлорэтилен |
4 |
1 |
|
Хлоропрен |
0,1 |
0,1 |
|
Четыреххлористый углерод |
4 |
2 |
ОПРЕДЕЛЕНИЕ ХЛОРОРГАНИЧЕСКИХ СОЕДИНЕНИЙ МЕТОДОМ СЖИГАНИЯ
В ПРИБОРЕ НИИ ГИГИЕНЫ ИМ Ф.Ф. ЭРИСМАНА [8]
Принцип определения
Хлорированные углеводороды сжигают в колонке с платиновой проволокой, накаленной докрасна (600 — 700 °C). Продукты сжигания поглощают раствором щелочи, в которой определяют ион хлора.
Чувствительность определения по хлору 1 мкг в анализируемом объеме. Хлориды, бромиды и йодиды мешают определению.
Аппаратура
Прибор для сжигания хлорированных соединений смонтирован на деревянной доске (схема показана на рис. 24). Состоит из двух частей: очистительной и аналитической. Аналитическая часть в свою очередь состоит из газовой пипетки емкостью 500 мл (7), стеклянной колонки для сжигания (2), в которую вмонтирована платиновая спираль (9) длиной 70 мм, сечением 0,3 мм, микропоглотителя (3) и уравнительной склянки на 550 мл (8). Микропоглотитель представляет собой пробирку высотой 45 — 50 мм, диаметром 12 мм (6), в которую вставлена стеклянная трубка (4) длиной 70 мм, диаметром 8 мм со шлифом в верхней части. Трубка соединена с колонкой сожжения, нижняя часть трубки оттянута и имеет диаметр 3 мм. В эту трубку помещена спираль из стеклянной палочки в 20 витков (5), длина спирали 50 — 52 мм, диаметр 7,5 мм. Спираль должна плотно входить в трубку. Это важно для полноты поглощения продуктов сожжения пробы.

Рис. 24. Схема прибора для определения
хлорированных углеводородов
Газовая пипетка (7) соединяется с колонкой сожжения посредством трехходового крана, который расположен вверху над пипеткой. Посредством этого крана можно соединить колонку с очистительной системой. На обратной стороне доски помещена очистительная система (1), состоящая из двух поглотительных приборов, первый прибор наполнен 5% раствором щелочи для поглощения хлористого водорода. Второй прибор содержит 0,01 н. раствор мышьяковистой кислоты для задержания хлора; газовая пипетка емкостью 500 мл. Она соединена с очистительной системой и посредством трехходового крана с колонкой сожжения; трансформатор мощностью 40 Вт.
Примечание. 1. Новый прибор, а также прибор после продолжительного употребления и после длительного перерыва в работе требует пропаривания. Для этого к прибору присоединяют парообразователь на 20 — 30 мин. После пропарки прибор осушают воздухом, не содержащим хлористого водорода и хлора. Колонку с платиновой спиралью включают в сеть только после того, как она высохнет. 2. Прибор для сжигания хлорированных углеводородов ПСУ изготавливают по МРТУ 42-2060-62 завод «Химлаборпосуда» (рис. 25).
Рис. 25. Прибор ПСУ, изготовленный по МРТУ 42-2060-62
А — обратная сторона прибора; Б — общий вид
Реактивы
Все реактивы готовят на дважды перегнанной воде, свободной от иона хлора.
1. Поглотительный раствор — 0,01 н. раствор едкого натра.
2. Стандартный раствор: исходный раствор с содержанием 100 мкг/мл иона хлора, получают растворением навески хлорида калия. 0,0204 г соли растворяют в мерной колбе емкостью 100 мл. Рабочий стандартный раствор с содержанием 10 мкг/мл получают разбавлением исходного раствора в 10 раз.
3. Азотная кислота, уд. вес 1,34 — 1,4.
4. Нитрат серебра, 0,1% раствор: навеску 0,1 г нитрата серебра растворяют в 90 мл воды и в раствор вливают 10 мл азотной кислоты уд. веса 1,34 — 1,4.
5. Едкий натр, 5% и 10% растворы.
6. Сульфат натрия, насыщенный раствор.
7. Мышьяковистая кислота, 0,01 н. раствор 0,124 г мышьяковистого ангидрида растворяют в 1 мл 10% едкого натра, добавляют 200 мл воды и нейтрализуют осторожно 5% раствором серной кислоты в присутствии фенолфталеина. Раствор вливают в мерную колбу емкостью 250 мл. Объем раствора доводят до метки 5% раствором бикарбоната натрия.
8. Серная кислота, 5% раствор.
9. Бикарбонат натрия, 5% раствор.
Отбор пробы
Для определения разовой концентрации отбирают две пробы в газовые пипетки емкостью 500 — 1000 мл, предварительно наполненные насыщенным раствором сернокислого натрия. В месте отбора проб раствор выливают из пипеток в течение 5 — 10 мин. и газовая пипетка заполняется исследуемым воздухом. По окончании отбора проб зажимы на пипетках закрывают и в концы резиновых трубок вставляют стеклянные палочки.
Для определения среднесуточной концентрации отбирают пробу в течение суток при помощи аспиратора емкостью 3 л, заполненного насыщенным раствором сернокислого натрия. Скорость отбора 0,1 л/ч.
Подготовка прибора к анализу
Подготовка прибора к анализу состоит из следующих процессов: 1) промывка микропоглотителя, 2) проверка герметичности прибора, 3) продувание прибора воздухом, свободным от хлористого водорода и хлора, 4) контрольный анализ, т.е. проверка на чистоту прибора.
1. Микропоглотитель промывают хромовой смесью, затем водой и пропаривают его в течение 10 — 15 мин. После этого проверяют чистоту микропоглотителя на отсутствие иона хлора нитратом серебра в присутствии кислоты (азотной или серной). Если промывная вода помутнеет от нитрата серебра, микропоглотитель следует промывать и кипятить в воде и вновь проверить реакцию на ион хлора. Чистый микропоглотитель присоединяется к колонке для сожжения.
2. Проверка герметичности прибора заключается в том, что уравнительные склянки заполняют дистиллированной водой, помещают на верхнюю полку прибора, все краны и зажимы открывают, но предварительно на нижний конец микропоглотителя надевают резиновую трубку с плотно завернутым зажимом и с вставленной стеклянной палочкой на конце трубки.
Прибор можно считать герметичным, если вода из уравнительной склянки не поступает в пипетку.
3. Для продувания прибора воздухом, свободным от хлора и хлористого водорода, необходимо: А) заполнить газовую пипетку, помещенную на обратной стороне прибора, водой, а затем чистым воздухом продуть прибор; Б) заполнить газовые пипетки, помещенные на лицевой стороне прибора, водой, а затем очищенным воздухом.
Продуть прибор чистым воздухом.
А. При заполнении пипетки, помещенной на обратной стороне прибора, соединяют нижний конец ее с уравнительной склянкой, а верхний конец пипетки соединяют с помощью трехходового крана с колонкой сожжения. Уравнительную склянку помещают на верхнюю полку прибора. Осторожно открывают кран у нижнего конца пипетки, и вода поступает в пипетку, а воздух из пипеток выходит через колонку сожжения в помещение лаборатории.
При наполнении пипетки водой зажим у нижнего края закрывают и для заполнения ее воздухом уравнительную склянку помещают на стол, трехходовым краном пипетку соединяют с очистительной системой. Нижний зажим на пипетке открывают, вода вытекает из пипетки в уравнительную склянку, а воздух через очистительную систему наполняет пипетку.
Б. Заполняют водой пипетку, помещенную на лицевой стороне прибора. Уравнительные склянки соединяют с нижними концами пипеток, зажимы должны быть закрыты, помещают их на верхнюю полку. Трехходовыми кранами пипетки соединяют с колонками сожжения. Осторожно открывают зажимы у нижних концов пипеток, вода поступает в пипетки, а воздух через колонки выходит в помещение лаборатории. После наполнения пипеток водой нижние зажимы закрывают, уравнительные склянки помещают на стол, трехходовые краны повертывают и соединяют пипетки с очистительной системой. Зажимы у нижних концов пипетки открывают, вода постепенно вытекает в уравнительные склянки и воздух через очистительную систему наполняет пипетки. По окончании наполнения трехходовые краны повертывают на нейтральное положение.
В. После этого требуется продуть прибор чистым воздухом. Для этого уравнительные склянки помещают на верхние полки прибора. Пипетки, помещенные на лицевой стороне прибора, соединяют с колонками сожжения трехходовыми кранами. Нижние зажимы у пипеток осторожно открывают и вода поступает в пипетки, а воздух, проходя колонки сожжения и микропоглотитель, выходит в помещение лаборатории. Необходимо промыть прибор чистым воздухом с помощью пипетки, помещенной на обратной стороне прибора. После 2 — 3-кратного промывания проводят контрольный опыт.
4. При контрольном анализе, прибор заполнен чистым воздухом, газовую пипетку заполняют чистым воздухом, как описано выше. Микропоглотители снимают с прибора и заполняют 0,01 н раствором едкого натра, вливая 3 — 4 капли его на внутреннюю спираль микропоглотителя и 2 — 3 капли — в пробирку поглотителя (нижний конец внутренней трубки должен быть погружен в раствор). Микропоглотитель соединяют с колонками сожжения. Газовые пипетки на лицевой стороне прибора соединяют внизу с уравнительными склянками, заполненными насыщенным раствором сульфата натрия. Верхние концы пипеток соединяют с трехходовыми кранами встык. Кранами пипетки соединяют с колонками сожжения. Колонки включают в сеть, при этом платиновая спираль быстро нагревается до красного каления.
Уравнительные склянки помещают на верхнюю полку прибора и открывают сначала верхние зажимы газовых пипеток, затем нижние.
Раствор из уравнительных склянок поступает в пипетки, а воздух проходит через колонки сожжения и микропоглотители. Прохождение воздуха наблюдается в микропоглотителе в виде цепочки пузырьков воздуха. Скорость потока воздуха 20 — 25 мл/мин. После вытеснения воздуха из пипеток трехходовой кран повертывают на нейтральное положение, закрывают зажимы на пипетках, уравнительные склянки ставят на стол и соединяют с нижним концом газовой пипетки, помещенной на обратной стороне прибора.
Уравнительную склянку помещают на верхнюю полку и трехходовым краном соединяют пипетку с колонкой сожжения и осторожно открывают нижний зажим у пипетки. Раствор поступает в пипетку; воздухом промывают прибор в течение 5 — 10 мин.
По окончании промывки прибора выключают колонки из сети, отъединяют микропоглотители от колонок, промывают их поглотительным раствором. В перпендикулярно поставленный микропоглотитель вносят каждый раз по 5 — 6 капель раствора. Раствор из поглотителя переливают в колориметрические пробирки с меткой 1 мл. Операцию промывания повторяют несколько раз и промывной раствор сливают в 2 — 3 пробирки. Количество раствора в каждой пробирке должно быть 1 мл.
В пробирки вносят по 0,2 мл 0,1% раствора нитрата серебра в азотной кислоте. При отрицательной реакции на хлор прибор можно считать чистым и пригодным для проведения анализа. В противном случае контрольный анализ повторяют несколько раз.
Ход анализа
В микропоглотитель вносят 3 — 4 капли 0,01 н. раствора едкого натра через верхнюю часть и 2 — 3 капли этого раствора помещают в пробирку микропоглотителя, последний соединяют с колонкой сожжения. Газовую пипетку с пробой присоединяют к трехходовому крану встык, она должна быть в вертикальном положении. Нижний конец пипетки соединяют с уравнительной склянкой. Трехходовой кран подвертывают так, чтобы пипетка с пробой была соединена с колонкой сожжения. Колонку включают в сеть и нагревают до красного каления и после этого открывают верхний зажим пипетки с пробой, затем открывают нижний зажим у пипетки, воздух из пипетки пропускают со скоростью 20 — 25 мл/мин. Далее поступают так, как указано в контрольном анализе.
После промывания микропоглотителя в колориметрические пробирки наливают по 0,2 мл 0,1% раствора нитрата серебра и помутнение проб сравнивают через 5 мин. после вливания нитрата со шкалой стандартов, которую готовят одновременно с пробой (табл. 48).
Таблица 48
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
|
Рабочий стандартный раствор, мл |
0 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
|
Поглотительный раствор, мл |
1 |
0,9 |
0,8 |
0,6 |
0,4 |
0,2 |
0 |
|
Содержание хлора, мкг |
0 |
1 |
2 |
4 |
6 |
8 |
10 |
Все пробирки шкалы обрабатывают аналогично пробе.
Расчет концентраций хлорорганических соединений (мг/м3) воздуха проводят по формуле:
![]()
где: a — количество хлора в пробе (мкг); к — коэффициент пересчета хлора на искомое соединение; V0 — объем воздуха в литрах, отобранного для анализа, приведенного к нормальным условиям.
Примечания. 1. Для нижеуказанных веществ даны коэффициенты пересчета с хлора на: дихлорэтан — 1,4, четыреххлористый углерод — 1,08, хлороформ — 1,13, трихларэтилен — 1,23, тетрахлорэтан — 1,01, хлоропрен — 2,49.
2. В случае необходимости определения других хлорорганических веществ коэффициент пересчета устанавливают в каждом случае отдельно, сжигая известные количества вещества, внесенные в специальный «гусек».
МЕТА- И ПАРАХЛОРАНИЛИНЫ
Метахлоранилин — жидкость, не растворимая в воде, температура кипения 230 °C, плотность при 20 °C 1,216, хорошо растворима в этаноле и эфире. Оба изомера обладают запахом, напоминающим нафталин.
Хлоранилины — вещества ядовитые, токсичные, их действие сходно с анилином.
Предельно допустимые концентрации парахлоранилина: максимальная разовая 0,04 мг/м3, среднесуточная 0,01 мг/м3.
Предельно допустимая концентрация метахлоранилина среднесуточная 0,01 мг/м3.
ОПРЕДЕЛЕНИЕ ПАРА- и МЕТАХЛОРАНИЛИНОВ
С ПАРАДИМЕТИЛАМИНОБЕНЗАЛЬДЕГИДОМ [3]
Принцип определения
При взаимодействии м- и п-хлоранилинов с парадиметиламинобензальдегидом в кислой среде образуется соединение, окрашенное в желтый цвет.
Чувствительность определения 0,25 мкг в анализируемом объеме раствора. Непредельные углеводороды, высшие спирты, амины мешают определению.
Реактивы
1. Поглотительный раствор — 10% раствор уксусной кислоты.
2. Стандартный раствор хлоранилина. Готовят исходный раствор в мерной колбе емкостью 50 мл, в которую вливают 15 — 20 мл поглотительного раствора. Колбу взвешивают и в нее вносят несколько кристаллов или капель хлоранилина свежеперекристаллизованного или перегнанного и вновь взвешивают. По разности двух взвешиваний устанавливают количество хлоранилина в колбе и рассчитывают его содержание в 1 мл раствора.
Рабочий стандартный раствор с содержанием 10 мкг/мл получают в мерной колбе емкостью 50 мл, в нее вливают такое количество исходного раствора, которое содержит 500 мкг хлоранилина, и объем жидкости доводят до метки поглотительным раствором.
3. Парадиметиламинобензальдегид, 2% раствор в 40% растворе уксусной кислоты.
4. Уксусная кислота, 40% раствор.
Отбор пробы
а) Для определения разовой концентрации воздух протягивают со скоростью 0,5 л/мин. через два соединенных поглотительных прибора с пористыми фильтрами N 1, наполненных по 3 мл поглотительного раствора в каждом. Продолжительность отбора 20 — 30 мин. при охлаждении.
б) Для определения среднесуточной концентрации воздух протягивают со скоростью 0,3 л/мин. через поглотительные приборы непрерывно в течение суток. Допустимо отбор проб проводить 6 — 12 раз с перерывами 4 — 2 ч в одни и те же поглотительные приборы.
Ход анализа
Анализируют пробу из каждого поглотительного прибора отдельно.
2 мл пробы переносят в пробирки, вливают по 0,6 мл 25% раствора парадиметиламинобензальдегида, содержимое пробирок взбалтывают и через 5 мин. измеряют оптическую плотность при длине волны 445 нм в кювете с толщиной слоя 0,5 см по сравнению с контролем, который готовят одновременно и аналогично пробе.
Содержание хлоранилина определяют по предварительно построенному калибровочному графику. Готовят шкалу стандартов (табл. 49).
Таблица 49
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
|
Рабочий стандартный раствор, мл |
0 |
0,025 |
0,05 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
|
Поглотительный раствор, мл |
2 |
1,975 |
1,95 |
1,9 |
1,8 |
1,6 |
1,4 |
1,2 |
1,0 |
|
Содержание хлоранилина, мкг |
0 |
0,25 |
0,5 |
1,0 |
2,0 |
4,0 |
6,0 |
8,0 |
10,0 |
Все пробирки шкалы обрабатывают аналогично пробе.
Шкалой стандартов можно пользоваться для визуального определения, ее готовят в колориметрических пробирках одновременно с пробами.
Расчет см. выше.
ХЛОРБЕНЗОЛ
Бесцветная жидкость с температурой кипения 132°, плотностью при 20 °C 1,106, мало растворимая в воде, но хорошо растворимая в стироле и других органических растворителях.
Хлорбензол — наркотик, действует на кроветворные органы, но слабее, чем бензол.
Предельно допустимые концентрации: максимальная разовая и среднесуточная 0,10 мг/м3.
ОПРЕДЕЛЕНИЕ ХЛОРБЕНЗОЛА НИТРОВАНИЕМ [1]
Принцип определения
При нитровании хлорбензола образуется нитросоединение, которое в щелочной среде в присутствии кетона дает соединение, окрашенное в розовый цвет.
Чувствительность определения — 1 мкг в анализируемом объеме раствора.
Ароматические соединения мешают определению.
Реактивы
1. Стандартный раствор хлорбензола: готовят исходный раствор в мерной колбе емкостью 50 мл, в которую наливают 2 — 3 мл нитрационной смеси, и колбу взвешивают. Затем в колбу вносят 2 — 3 капли хлорбензола, вновь взвешивают, раствор встряхивают и помещают колбу в кипящую водяную баню на 40 мин. После охлаждения раствор доводят до метки нитрационной смесью. По разности двух взвешиваний устанавливают количество хлорбензола в 1 мл раствора.
Рабочий стандартный раствор с содержанием 100 мкг/мл готовят соответствующим разведением исходного раствора.
Другие реактивы и отбор пробы см. «Определение бензола нитрованием» выше.
Ход анализа
Поглотительный прибор с пробой помещают в кипящую водяную баню на 40 мин. По охлаждении раствор переводят в коническую колбу емкостью 50 мл и поглотительный прибор ополаскивают несколько раз 5 — 6 мл воды. Промывную воду присоединяют к пробе, избегая сильного разогревания раствора. После охлаждения раствор нейтрализуют 25% раствором аммиака. Нейтрализацию проводят, пользуясь бюреткой с краном в присутствии лакмусовой бумаги. Затем пробу переносят в делительную воронку и добавляют 2,2 мл бутанона, смесь в воронке встряхивают в течение 5 мин. до разделения слоев. Бутаноновый раствор переливают и объем доводят бутаноном до 2 мл, прибавляют 0,75 мл щелочи. Содержимое пробирки встряхивают в течение нескольких минут и через 20 — 30 мин. сравнивают интенсивность пробы со шкалой стандартов. Одновременно готовят раствор для стандартной шкалы. Для этого берут 1 мл рабочего стандартного раствора, выливают в коническую колбу емкостью 100 мл, в которую внесено 8 — 10 мл воды. Раствор охлаждают и нейтрализуют 25% раствором аммиака в присутствии лакмуса.
Охлажденный нейтральный раствор выливают в делительную воронку и добавляют 10 мл бутанона. Раствор встряхивают в течение 10 мин. и оставляют в покое до полного разделения слоев. Нижний слой, водный, сливают через кран, верхний, бутаноновый, переливают через верх воронки в мерную колбу емкостью 10 мл или в пробирку с притертой пробкой и меткой 10 мл. Объем раствора доводят до метки бутаноном. Получают стандартный раствор в бутаноне с содержанием 10 мкг/мл. Из этого раствора готовят шкалу стандартов (табл. 50).
Таблица 50
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
|
Стандартный бутаноновый раствор, мл |
0 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
|
Бутанон, мл |
2 |
1,9 |
1,8 |
1,6 |
1,4 |
1,2 |
1,0 |
|
Содержание хлорбензола, мкг |
0 |
1 |
2 |
4 |
6 |
8 |
10 |
Все пробирки шкалы обрабатывают аналогично пробе.
Расчет см. выше.
НИТРОХЛОРБЕНЗОЛЫ
Нитрохлорбензолы орто-, мета-, пара-; представляют собой кристаллические вещества с запахом нитробензола.
Относятся к токсическим соединениям.
Оказывают действие на нервную систему, печень и др. Действие их сходно с действием нитробензола.
Предельно допустимые концентрации пара- и ортонитробензолов; среднесуточная 0,004 мг/м3.
ОПРЕДЕЛЕНИЕ МЕТА-, ПАРАНИТРОХЛОРБЕНЗОЛА
С ПАРАДИМЕТИЛАМИНОБЕНЗАЛЬДЕГИДОМ [4]
Принцип определения
Метод основан на восстановлении нитрохлорбензола до хлоранилина, который при взаимодействии с п-диметиламинобензальдегидом образует соединение, окрашенное в желтый цвет.
Чувствительность определения — 0,2 мкг в анализируемом объеме раствора.
Анилин, хлоранилин, дихлоранилин и другие ароматические амины мешают определению, а также спирты и альдегиды.
Реактивы, отбор пробы, ход анализа см. «Определение хлоранилина с парадиметиламинобензальдегидом» выше.
МЕТА- И ПАРАХЛОРФЕНИЛИЗОЦИАНАТЫ
|
ClC6H4(NCO) |
Мол. вес 153,58 |
Метахлорфенилизоцианат — бесцветная жидкость, не растворима в воде, слабо растворима в водных растворах щелочи.
Парахлорфенилизоцианат — твердое кристаллическое вещество белого цвета, очень гигроскопично, обладает резким запахом, в воде не растворимо и трудно растворимо в водных растворах щелочи.
Хлорфенилизоцианаты токсичны.
Предельно допустимые концентрации: максимальная разовая и среднесуточная для парахлорфенилизоцианата 0,0015 мг/м3, для метахлорфенилизоцианата 0,005 мг/м3.
ОПРЕДЕЛЕНИЕ МЕТА- и ПАРАХЛОРФЕНИЛИЗОЦИАНАТОВ
ПО ОБРАЗОВАНИЮ ХЛОРАНИЛИНА [5]
Принцип определения
При взаимодействии со щелочью мета- и парахлорфенилизоцианат гидролизуется с образованием хлоранилина, который диазотируют и сочетают с ![]() -нафталом, при этом образуется продукт, окрашенный в розовый цвет.
-нафталом, при этом образуется продукт, окрашенный в розовый цвет.
Чувствительность определения — 0,1 мкг в анализируемом объеме раствора.
Анилин, мета- и парахлоранилин, фенилизоцианат мешают определению.
Реактивы
1. Поглотительный раствор — 0,5 н. раствор едкого натра.
2. Стандартный раствор мета- или парахлорфенилизоцианата. Готовят исходный раствор, для этого в мерную колбу емкостью 25 — 50 мл наливают 15 — 20 мл 0,5 н. раствора едкого натра, взвешивают, вносят в нее несколько кристалликов продукта, снова взвешивают. По разности двух взвешиваний устанавливают количество хлорфенилизоцианата в 1 мл раствора.
Рабочий стандартный раствор с содержанием 10 мкг/мл готовят путем соответствующего разведения исходного раствора.
Раствор применяют свежеприготовленный.
3. Аммиак, 0,5% раствор.
4. Уксусная кислота, 1 н. раствор.
5. Нитрит натрия, 7% раствор.
6. Бромид натрия, 12% раствор.
7. Составной раствор: растворы нитрита натрия и бромида натрия смешивают в отношении 1:1.
8. Едкий натр, 20% раствор.
9. Этанол 96°.
10. ![]() -Нафтол, 0,05% раствор в этаноле.
-Нафтол, 0,05% раствор в этаноле.
Отбор пробы
Воздух со скоростью 1 л/мин. протягивают через два последовательно соединенных поглотительных прибора с пористым фильтром N 1, наполненных по 1,5 мл поглотительного раствора. Продолжительность отбора 20 — 30 мин. Анализ проб проводят в день отбора.
Ход анализа
Содержимое каждого поглотительного прибора анализируют отдельно. 1 мл пробы помещают в пробирку с притертой пробкой с меткой 5 — 10 мл. В пробирку с пробой наливают 1 мл 0,5% раствора аммиака, 0,9 мл 1 н. раствора уксусной кислоты, объем раствора в пробирке доводят до 5 мл водой, раствор встряхивают и добавляют 0,3 мл составного раствора. Через 5 мин. в пробирку наливают 0,1 мл 20% раствора едкого натра, раствор перемешивают и в него вливают 0,5 мл 0,05% раствора альфа-нафтола и 1 мл этанола. Содержимое пробирки встряхивают и оставляют в покое на 50 — 60 мин.
По истечении этого времени измеряют оптическую плотность раствора в кювете с толщиной слоя 1 см по сравнению с контролем, который готовят одновременно и аналогично пробе. Определение проводят при длине волны 508 нм.
Содержание м-п-хлорфенилизоцианата в анализируемом объеме определяют по предварительно построенному калибровочному графику. Для построения графика готовят шкалу стандартов (табл. 51).
Таблица 51
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
9 |
|
Рабочий стандартный раствор, мл |
0 |
0,01 |
0,03 |
0,05 |
0,07 |
0,1 |
0,3 |
0,5 |
0,7 |
1,0 |
|
Поглотительный раствор, мл |
1 |
0,99 |
0,97 |
0,95 |
0,93 |
0,9 |
0,7 |
0,5 |
0,8 |
0 |
|
Содержание МХФЦ или ПХФЦ, мкг |
0 |
0,1 |
0,3 |
0,5 |
0,7 |
1,0 |
3,0 |
5,0 |
7,0 |
10 |
Все пробирки обрабатывают аналогично пробам. Шкалой стандартов можно пользоваться для визуального определения, ее готовят в колориметрических пробирках одновременно с пробами.
Расчет см. выше.
ЭПИХЛОРГИДРИН
|
|
Мол. вес 117 |
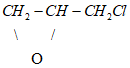
Прозрачная бесцветная жидкость, температура кипения 117 °C, плотность 1,18, мало растворима в воде, но хорошо растворима в этаноле и эфире.
Эпихлоргидрин действует раздражающе на слизистые оболочки дыхательных путей.
Предельно допустимые концентрации: максимальная разовая и среднесуточная 0,2 мг/м3.
ОПРЕДЕЛЕНИЕ ЭПИХЛОРГИДРИНА ПО ФОРМАЛЬДЕГИДУ [7, 10]
Принцип определения
При взаимодействии эпихлоргидрина с йодной кислотой в сернокислой среде образуется формальдегид, который определяют с хромотроповой кислотой.
Чувствительность определения — 0,5 мкг в анализируемом объеме раствора.
Формальдегид, окись этилена, этиленгликоль мешают определению.
Реактивы
1. Поглотительный раствор — серная кислота, 60% раствор (по весу).
2. Стандартный раствор эпихлоргидрина. Готовят исходный стандартный раствор: в мерную колбу емкостью 50 мл вливают 10 — 15 мл поглотительного раствора и взвешивают. Затем в колбу вносят 2 капли эпихлоргидрина и вновь взвешивают. По разности двух взвешиваний устанавливают количество эпихлоргидрина в 1 мл раствора.
Рабочий стандартный раствор с содержанием 10 мкг/мл эпихлоргидрина получают соответствующим разбавлением исходного.
3. Перйодат калия, 1% раствор в 60% серной кислоте.
4. Сульфит натрия, 40% раствор готовят при нагревании. Устойчив в течение 2 — 3 дней.
5. Хроматроповая кислота, 0,3 г кислоты растворяют в 5 мл воды, затем осторожно приливают 95 мл серной кислоты удельного веса 1,84. Раствор устойчив в течение недели при хранении в темном прохладном месте.
6. Силикагель ACM.
Отбор пробы
а) Для определения разовой концентрации протягивают воздух со скоростью 0,5 л/мин. через поглотительный прибор с пористым фильтром N 1, наполненный 6 мл поглотительного раствора.
При отборе проб на твердый сорбент по 2 мл силикагеля помещают в два поглотительных прибора Института имени Ф.Ф. Эрисмана. Отбор проводят в «кипящий» слой. Скорость отбора 5 л/мин.
б) Для определения среднесуточной концентрации воздух со скоростью 0,1 — 0,2 л/мин. протягивают через поглотительный прибор непрерывно в течение суток. В первом и втором случае отбор проводят в течение 20 — 30 мин.
Допустимо отбор проводить 6 — 12 раз с перерывами 4 — 2 ч по 20 — 30 мин. в один и тот же поглотительных прибор.
Отбор среднесуточных проб можно проводить в кипящий слой силикагеля, скорость до 5 л/мин.
Ход анализа
В пробирку помещают 5 мл пробы, в нее вносят 1 каплю перйодата калия, раствор перемешивают и пробирку помещают на 15 мин. в кипящую водяную баню. После охлаждения раствора прибавляют осторожно 5 капель сульфита и тщательно перемешивают. Раствор должен обесцветиться. Избытка сульфита необходимо избегать. Через 2 — 3 мин. в пробирку приливают 1 мл хромотроповой кислоты. Раствор перемешивают и помещают на 30 мин. в кипящую водяную баню. После охлаждения раствора в пробирку вливают 2 мл воды и через 20 — 30 мин. измеряют оптическую плотность в кювете толщиной слоя 1 см по сравнению с контролем, который готовят одновременно и аналогично пробе. Определение проводят при длине волны 570 нм.
Содержание эпихлоргидрина в анализируемом объеме определяют по предварительно построенному калибровочному графику. Для построения графика готовят шкалу стандартов (табл. 52).
Таблица 52
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
9 |
|
Рабочий стандартный раствор, мл |
0 |
0,05 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
1,2 |
1,6 |
2,0 |
|
Поглотительный раствор, мл |
5 |
4,95 |
4,9 |
4,8 |
4,6 |
4,4 |
4,2 |
3,8 |
3,4 |
3,0 |
|
Содержание эпихлоргидрина, мкг |
0 |
0,5 |
1 |
2 |
4 |
6 |
8 |
12 |
16 |
20 |
Все пробирки шкалы обрабатывают аналогично пробе. Шкалой стандартов можно пользоваться для визуального определения, ее готовят одновременно с пробой.
Ход анализа после отбора проб на силикагель. В каждую пробирку с пробой наливают по 4 мл поглотительного раствора и содержимое пробирок встряхивают. Затем готовят шкалу стандартов, как указано в табл. 52, но предварительно в каждую пробирку шкалы всыпают по 2 мл силикагеля. Вносят стандартный раствор и содержимое пробирок встряхивают. После этого объем в каждой пробирке доводят до 4 мл поглотительным раствором. Далее пробу и шкалу обрабатывают так, как при отборе пробы в жидкую поглотительную среду.
Расчет см. выше.
ЧЕТЫРЕХХЛОРИСТЫЙ УГЛЕРОД
Бесцветная, легко подвижная, летучая жидкость с неприятным запахом. Температура кипения 76,8°, плотность при 2° 1,595, мало растворима в воде, легко растворима в этаноле, эфире, ацетоне.
Четыреххлористый углерод является наркотиком, вызывает поражение печени, почек. Предельно допустимая максимальная разовая концентрация 4 мг/м3, среднесуточная 2 мг/м3.
ОПРЕДЕЛЕНИЕ ЧЕТЫРЕХХЛОРИСТОГО УГЛЕРОДА С ПИРИДИНОМ [6]
Принцип определения
При взаимодействии четыреххлористого углерода с пиридином образуется соединение, которое с анилином в присутствии ацетона дает дианилид глютаконового альдегида.
Чувствительность определения — 1 мкг в анализируемом объеме раствора. Хлороформ, хлораль и некоторые другие галоидоуглеводороды мешают определению. Хлористый метил, хлористый метилен, дихлорэтан, спирты, хлористый водород не мешают определению.
Реактивы
1. Поглотительный раствор — пиридин. Пиридин применяют свежеперегнанный, для этого его кипятят в течение часа с обратным холодильником над кристаллической щелочью (8 — 10 г на 100 мл).
Перегоняют, добавив кристаллической щелочи, и отбирают фракцию, кипящую при 114 — 116 °C. Хранят в стеклянной посуде в темном месте.
2. Стандартный раствор четыреххлористого углерода. Исходный стандартный раствор готовят растворением точной навески четыреххлористого углерода в пиридине.
Рабочий стандартный раствор с содержанием 10 мкг/мл готовят соответствующим разведением исходного раствора. Рабочий стандартный раствор устойчив в течение 6 ч.
3. Анилин, х.ч., свежеперегнанный.
4. Ацетон, х.ч.
5. Уксусная кислота, ледяная.
6. Натр едкий, 0,5 н. раствор.
Отбор пробы
Для определения разовой концентрации воздух со скоростью 0,1 л/мин. протягивают через два поглотительных прибора с пористым фильтром N 1, наполненные по 3 мл пиридина.
Продолжительность отбора 7 — 10 мин.
При наполнении приборов отмечают уровень жидкости и периодически доводят его до первоначального пиридином. Во время отбора поглотительные приборы охлаждают водой со льдом.
Ход анализа
Содержимое приборов количественно переносят в колориметрическую пробирку, добавляют 0,1 мл ацетона, 0,2 мл 0,5 н. раствора щелочи, перемешивают и нагревают в течение 4 мин. при 100 °C.
По охлаждении вносят 0,5 мл уксусной кислоты, 0,1 мл анилина и разбавляют смесь водой до 4 мл.
Через 15 мин. окрашенный в оранжевый цвет раствор фотометрируют при длине волны 485 — 495 нм в кювете с толщиной слоя 1 см по сравнению с контролем, который готовят одновременно и аналогично пробе. Содержание четыреххлористого углерода в анализируемом объеме определяют по предварительно калибровочному графику. Для построения графика готовят шкалу стандартов (табл. 53).
Таблица 53
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
|
Рабочий стандартный раствор, мл |
0 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
1 |
2 |
|
Поглотительный раствор, мл |
2 |
1,9 |
1,8 |
1,6 |
1,4 |
1,2 |
1 |
0 |
|
Содержание четыреххлористого углерода, мкг |
0 |
1 |
2 |
4 |
6 |
8 |
10 |
20 |
Все пробирки шкалы обрабатывают аналогично пробе. Шкалу стандартов можно использовать для визуального определения. Окраска растворов устойчива в течение 3 ч.
Расчет см. выше.
ХЛОРОФОС
|
|
Мол. вес 257,5 |
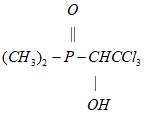
Твердое вещество с температурой плавления 81 °C, температурой кипения 120 °C (0,4 мм рт. ст.), легко растворимое в воде. Хлорофос используют как инсектофунгицид. Обладает токсическими свойствами.
Предельно допустимые концентрации: максимальная разовая 0,04 мг/м3, среднесуточная 0,02 мг/м3.
ОПРЕДЕЛЕНИЕ ХЛОРОФОСА ПО ХЛОРУ [9]
Принцип определения
Метод основан на омылении хлорофоса раствором щелочи при нагревании и определении иона хлора по реакции с нитратом серебра.
Чувствительность определения — 2 мкг хлорофоса в анализируемом объеме раствора. Определению мешают вещества, дающие реакцию с нитратом серебра.
Реактивы
1. Стандартный раствор с содержанием 100 мкг/мл хлорофоса. Готовят растворением 0,01 г хлорофоса в 100 мл бидистиллированной воды.
2. Едкий натр, 10% раствор.
3. Азотная кислота, 20% раствор.
4. Нитрат серебра, 1% раствор.
Все реактивы готовят на бидистиллированной воде.
Отбор пробы
Для определения разовой концентрации воздух со скоростью 3 — 4 л/мин. протягивают через систему, состоящую из патрона с фильтром АФА-XII и двух поглотительных приборов Рыхтера (малая модель), содержащих по 4 мл бидистиллированной воды. Продолжительность отбора 20 — 30 мин.
Ход анализа
Фильтр переносят в стакан и промывают 2 раза водой, доводят объем до 4 мл. Пробу из поглотительных приборов переносят в пробирки с притертыми пробками. К пробам приливают по 1 мл 10% раствора едкого натра и нагревают на кипящей водяной бане в течение 30 мин. По охлаждении прибавляют по 1 мл 10% раствора азотной кислоты и 0,1 мл 5% раствора нитрата серебра, тщательно взбалтывают и через 10 мин. нефелометрируют в кюветах с толщиной слоя 1 см при длине волны 364 нм по отношению к контролю, который строят одновременно и аналогично пробам.
Содержание хлорофоса в анализируемом объеме раствора определяют по предварительно построенному калибровочному графику.
Для построения калибровочного графика готовят шкалу стандартов (табл. 54).
Таблица 54
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
|
Рабочий стандартный раствор, мл |
0 |
0,02 |
0,05 |
0,1 |
0,2 |
0,3 |
0,4 |
0,5 |
|
Поглотительный раствор, мл |
2 |
1,98 |
1,95 |
1,9 |
1,8 |
1,7 |
1,6 |
1,5 |
|
Содержание хлорофоса, мкг |
0 |
2 |
5 |
10 |
20 |
30 |
40 |
50 |
Все пробирки шкалы обрабатывают аналогично пробам, измеряют оптическую плотность и строят график.
Шкалой стандартов можно пользоваться для визуального определения, ее готовят в колориметрических пробирках одновременно с пробами.
Расчет см. выше.
ХЛОРОПРЕН
(Хлористый дивинил, 2-хлор-1,3-бутадиен)
|
CH2=CH-CCl=CH2 |
Мол. вес 88,53 |
Бесцветная жидкость с температурой кипения 59,4 °C. Плотность 0,958 (20 °C), упругость пара 176,2 мм рт. ст. (20 °C). Хлоропрен легко полимеризуется, плохо растворим в воде, растворим в органических растворителях. Хлоропрен токсичен, раздражает дыхательные пути, вызывает дерматиты. Предельно допустимые концентрации: максимальная разовая и среднесуточная 0,10 мг/м3.
СПЕКТРОФОТОМЕТРИЧЕСКОЕ ОПРЕДЕЛЕНИЕ ХЛОРОПРЕНА [2]
Принцип определения
Метод основан на измерении оптической плотности спиртового раствора хлоропрена при длине волны 222,6 нм. Чувствительность определения — 0,5 мкг хлоропрена в анализируемом объеме раствора. Определению мешают вещества, поглощающие при длине волны 222,6 нм.
Реактивы
1. Поглотительный раствор — этиловый спирт 96°, ректификат.
2. Стандартные растворы хлоропрена. Исходный стандартный раствор хлоропрена готовят следующим образом. В мерную колбу емкостью 25 мл вносят 10 — 15 мл этилового спирта, взвешивают, затем вносят несколько капель свежеперегнанного хлоропрена и снова взвешивают. Объем в колбе доводят до метки спиртом, тщательно перемешивают и по разности между первым и вторым взвешиванием рассчитывают содержание хлоропрена в 1 мл раствора. Соответствующим разведением готовят стандартный раствор с содержанием 100 мкг/мл, а из него рабочий стандартный раствор с содержанием 10 мкг/мл хлоропрена. Растворы готовят в день анализа.
Отбор пробы
Для определения разовой концентрации воздух со скоростью 5 л/ч протягивают через 3 — 4 поглотительных прибора с пористой пластинкой N 1, содержащих по 10 мл этилового спирта. Во время отбора поглотительные приборы помещают в сосуд со льдом.
Ход анализа
Содержимое каждого поглотительного прибора переливают в кварцевую кювету с толщиной слоя 1 см и измеряют оптическую плотность на спектрофотометре при длине волны 222,6 нм.
В качестве контроля используют этиловый спирт. Содержание хлоропрена в анализируемом объеме определяют по предварительно построенному калибровочному графику. Для построения калибровочного графика готовят шкалу стандартов (табл. 55).
Таблица 55
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
|
Рабочий стандартный раствор, мл |
0 |
0,05 |
0,1 |
0,2 |
0,4 |
0,8 |
1,6 |
3,2 |
|
Этиловый спирт, мл |
10 |
9,95 |
9,9 |
9,8 |
9,6 |
9,2 |
8,4 |
6,8 |
|
Содержание хлоропрена, мкг |
0 |
0,5 |
1,0 |
2,0 |
4,0 |
8,0 |
16,0 |
32,0 |
Растворы шкалы стандартов фотометрируют в тех же условиях, что и пробы.
Расчет см. выше.
ЛИТЕРАТУРА
1. Алексеева М.В. Определение хлорбензола. В кн.: Определение вредных веществ в воздухе промышленных помещений. М., Госхимиздат, 1954.
2. Акопян И.Х., Абешян М.М. и др. Спектрофотометрический метод определения хлоропрена в атмосферном воздухе. Гиг. и сан., 1970, 9.
3. Андреещева Н.Г. Простые методы определения микроконцентраций анилина, метахлоранилина, парахлоранилина и 3,4-дихлоранилина в атмосферном воздухе. Гиг. и сан., 1968, 11.
4. Андреещева Н.Г. Определение микроконцентраций метанитрохлорбензола, п-нитрохлорбензола и нитробензола в атмосферном воздухе. Гиг. и сан., 1968, 12.
5. Беляков А.А., Тубина А.Я. Определение мета- и парахлорфенилизоционата. В сб.: Новое в области промышленно-санитарной химии. М., «Медицина», 1969.
6. Беляков А.А. Определение четыреххлористого углерода. В кн.: Определение вредных веществ в воздухе производственных помещений, г. Горький, 1970.
7. Гронсберг Е.Ш. Определение эпихлоргидрина. В сб.: Технические условия на методы определения вредных веществ в воздухе. М., 1964, в. 3.
8. Сендерихина Д.П. Определение хлорированных углеводородов в воздухе методом микросжигания. Гиг. и сан., 1954, 8.
9. Табакова С.А. Экспериментальные данные к гигиеническому нормированию хлорофоса. Гиг. и сан., 1969, 11.
10. Фомин А.П. Биологическое действие эпихлоргидрина и его гигиеническое значение как фактора загрязнения атмосферы. Гиг. и сан., 1966, 9.
Глава X
СПИРТЫ, ФЕНОЛЫ, ЭФИРЫ
СПИРТЫ ЖИРНОГО РЯДА
Спирты одноатомные насыщенные.
Физические свойства и предельно допустимые концентрации некоторых спиртов жирного ряда приведены в табл. 56.
Таблица 56
|
Наименование спирта |
Формула |
Молекулярный вес |
Состояние в обычных условиях |
Удельный вес (20°) |
Точка кипения °C |
ПДК, мг/м3 |
|
|
максимальная разовая |
среднесуточная |
||||||
|
Метиловый |
CH3OH |
32,04 |
Жидкость |
0,792 |
66 |
1,0 |
0,5 |
|
Этиловый |
C2H5OH |
46,03 |
То же |
0,789 |
78,4 |
5,0 |
5,0 |
|
Пропиловый |
C3H7OH |
60,09 |
« |
0,804 |
96 — 98 |
0,3 |
0,3 |
|
Изопропиловый |
C3H7OH |
60,09 |
« |
0,786 |
79,5 — 81,5 |
0,6 |
0,6 |
|
Бутиловый |
C4H9OH |
74,12 |
« |
0,810 |
118 |
0,1 |
0,1 |
|
Амиловый |
C5H11OH |
88,15 |
« |
0,802 |
137,7 |
— |
— |
|
Изооктиловый |
C8H17OH |
130,23 |
« |
0,827 |
194 |
0,15 |
0,15 |
Одноатомные насыщенные спирты обладают выраженными наркотическими свойствами. Сила наркотического действия возрастает с увеличением молекулярного веса. Пары спиртов обладают также раздражающим действием на слизистые оболочки.
Метиловый спирт занимает особое место среди спиртов этого ряда, так как в организме он дает весьма токсичные соединения. Это сильный нервный и сосудистый яд, обладающий кумулятивным действием. Для него весьма характерным является поражение зрительного нерва и сетчатки глаза.
МЕТИЛОВЫЙ СПИРТ (МЕТАНОЛ)
ОПРЕДЕЛЕНИЕ МЕТИЛОВОГО СПИРТА ПО ФОРМАЛЬДЕГИДУ [2]
Принцип определения
Метод основан на окислении метилового спирта до формальдегида, который определяют по реакции с хромотроповой кислотой.
Чувствительность фотометрического определения — 4 мкг метилового спирта в анализируемом объеме раствора, чувствительность визуального определения — 0,4 мкг.
Формальдегид мешает определению.
Реактивы
1. Поглотительный раствор: дважды перегнанная вода.
2. Стандартные растворы метилового спирта. Исходный раствор готовят так: в мерную колбу емкостью 50 мл вливают 10 мл дважды перегнанной воды и взвешивают на аналитических весах. Затем вносят в колбу 2 — 3 капли метанола и снова взвешивают, по разности весов определяют количество метанола. Раствор в колбе доводят до метки водой. Вычисляют содержание метанола в 1 мл раствора. Из этого раствора готовят раствор с содержанием 10 мг/мл. Перед определением готовят рабочий стандартный раствор с содержанием в 1 мл 10 мкг метанола.
3. Серная кислота 9 М. К 530 мл дистиллированной воды осторожно приливают 470 мл серной кислоты удельного веса 1,82.
4. Серная кислота 15 М. К 100 мл воды осторожно приливают 900 мл серной кислоты удельного веса 1,80 — 1,82.
5. Перманганат калия, 1% раствор.
6. Хромотроповая кислота (1,8-диоксинафталин, 3,6-дисульфокислота). 0,5 г хромотроповой кислоты растворяют в 10 мл воды, раствор применяют свежеприготовленным. Для определения готовят следующий раствор: берут 2 мл раствора хромотроповой кислоты в колбу емкостью 50 мл и доводят раствор до метки 15 М серной кислотой.
7. Сульфит натрия, 3% раствор, применяют свежеприготовленным.
8. Фосфорная кислота, 85% раствор, уд. вес 1,71.
Отбор пробы
а) Для определения разовой концентрации воздух со скоростью 0,5 л/мин. протягивают через два поглотительных прибора с пористой пластинкой N 1, содержащих по 5 мл дважды перегнанной воды. Продолжительность отбора 20 — 30 мин.
Ход анализа
Из каждого поглотительного прибора переносят в пробирку 2 мл пробы, добавляют по 0,25 мл фосфорной кислоты, а затем по 2 капли 1% раствора перманганата калия и помещают точно на 15 мин. в баню со льдом. По истечении этого времени пробирки вынимают из бани и в них по каплям вносят 3% раствор сульфита, до обесцвечивания раствора, избегая избытка сульфита. Затем во все пробирки вливают по 2 мл хромотроповой кислоты и помещают их на 30 мин. в кипящую водяную баню. По охлаждении в пробирки наливают 9 М серную кислоту до объема 5 мл, перемешивают и через 50 — 60 мин. фотометрируют в кювете с толщиной слоя 1 см при длине волны 574 нм по сравнению с контролем, который готовят одновременно и аналогично пробе. Содержание метанола в анализируемом объеме определяют по предварительно построенному калибровочному графику. Для построения калибровочного графика готовят шкалу стандартов (табл. 57).
Таблица 57
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
|
Рабочий стандартный раствор, мл |
0 |
0,4 |
0,6 |
0,8 |
1,0 |
2,0 |
|
Вода, мл |
2 |
1,6 |
1,4 |
1,2 |
1,0 |
0 |
|
Содержание метанола, мкг |
0 |
4 |
6 |
8 |
10 |
20 |
Все пробирки шкалы обрабатывают аналогично пробам и строят калибровочный график.
Шкалой стандартов можно пользоваться для визуального определения, ее готовят в колориметрических пробирках одновременно с пробами (табл. 58).
Таблица 58
ШКАЛА СТАНДАРТОВ ДЛЯ ВИЗУАЛЬНОГО ОПРЕДЕЛЕНИЯ МЕТАНОЛА
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
|
Рабочий стандартный раствор, мл |
0 |
0,04 |
0,06 |
0,08 |
0,1 |
0,2 |
|
Вода, мл |
2 |
1,96 |
1,94 |
1,92 |
1,9 |
1,8 |
|
Содержание метанола, мкг |
0 |
0,4 |
0,6 |
0,8 |
1,0 |
2,0 |
Расчет см. выше.
———————————
<1> Раздельное определение метилового спирта и формальдегида см. ниже.
ОПРЕДЕЛЕНИЕ ОБЩЕГО КОЛИЧЕСТВА СПИРТОВ
ГРУППЫ C1 — C10 С ВАНАДИЙ-ОКСИХИНОЛИНОВЫМ КОМПЛЕКСОМ
(МЕТИЛОВОГО, ЭТИЛОВОГО, ПРОПИЛОВОГО, БУТИЛОВОГО,
АМИЛОВОГО, ГЕКСИЛОВОГО, ГЕПТИЛОВОГО, ОКТИЛОВОГО,
НОНИЛОВОГО, ДЕЦИЛОВОГО) [8]
Принцип определения
При взаимодействии спиртов с ванадий-оксихинолиновым комплексом образуется соединение, окрашенное в розово-оранжевый цвет. Избыток ванадий-оксихинолинового комплекса, окрашенный в черный цвет, разрушают щелочью.
Чувствительность — 5 мкг спирта в определяемом объеме раствора.
Не мешают определению ацетон, формальдегид, динил, кислоты жирного ряда (муравьиная, уксусная, масляная и др.), бутилацетат, метилацетат, винилацетат, сложные эфиры ортофосфорной кислоты (бутиловый, диметиловый, гептиловый, октиловый, нониловый) в количестве до 2 мг.
Реактивы
1. Стандартные растворы отдельных спиртов. Исходные стандартные растворы с содержанием 1 мг/мл готовят растворением расчетных количеств спиртов в бензоле. Рабочие стандартные растворы с содержанием 100 мкг спирта в 1 мл готовят разбавлением исходных растворов в 10 раз бензолом. Стандартные растворы сохраняются в течение суток. Для приготовления стандартного раствора используют спирт, который содержится в воздухе в большем количестве или наиболее токсичен.
2. Бензол х.ч. Очищают бензол следующим образом: к 900 мл бензола добавляют 100 мл серной кислоты уд. веса 1,84, помещают смесь в делительную воронку и встряхивают. Повторяют операцию до тех пор, пока кислота не перестанет окрашиваться или будет иметь слабо желтую окраску. После удаления кислоты бензол дважды встряхивают с водой, затем с 10% раствором едкого натра и снова с водой до нейтральной реакции (по фенолфталеину). Очищенный бензол осушают прокаленным хлористым кальцием в течение 1 — 2 сут., фильтруют и хранят в закрытой склянке.
3. Уксусная кислота, 5% раствор.
4. 8-оксихинолин, 2% раствор в 5% растворе уксусной кислоты.
5. Ванадат аммония, 0,08% раствор.
6. Едкий натр, 1 н. раствор.
Реактивы устойчивы в течение 3 мес.
7. Ванадий-оксихинолиновый комплекс. Готовят непосредственно перед использованием. В делительную воронку вносят 2,4 мл 5% раствора уксусной кислоты, 1,2 мл раствора 8-оксихинолина, 4 мл раствора ванадата аммония. Содержимое воронки перемешивают, затем добавляют 8 мл бензола и встряхивают смесь растворов в течение 0,5 мин. После разделения слоев нижний, водный, удаляют. Верхний слой, представляющий собой раствор ванадий-оксихинолинового комплекса в бензоле, сливают в сухую пробирку с пробкой.
8. Активированный уголь марки АГ-5 обрабатывают концентрированной соляной кислотой в течение 1 — 1 1/2 г ч при кипячении. Затем кислоту сливают, а уголь промывают несколько раз дистиллированной водой, 0,1 н. раствором аммиака до тех пор, пока последний перестанет окрашиваться. Далее уголь снова промывают дистиллированной водой до отрицательной реакции на ион хлора и сушат тонким слоем при 100 — 120 °C в течение 1 — 2 ч. Хранят уголь в склянке с притертой пробкой.
Отбор пробы
а) Для определения разовой концентрации воздух со скоростью 1 л/мин. протягивают через поглотительную трубку (см. рис. 6), наполненную 1 г угля. Поглотительная трубка с двух сторон закрывается спиралью из проволоки во избежание высыпания угля. Продолжительность отбора не менее 50 мин.
б) Для определения среднесуточной концентрации воздух со скоростью до 1 л/мин. протягивают через трубку, наполненную 1 г угля. Отбор проб проводят непрерывно в течение суток. Допустимо отбор проб проводить в одну и ту же трубку 6 — 12 раз с перерывами в 4 или 2 ч.
Ход анализа
Уголь переносят в пробирку с притертой пробкой, добавляют 4 мл бензола. Содержимое пробирки встряхивают в течение 2 — 3 мин. и нагревают в течение часа при 55 — 60 °C, затем вновь встряхивают в течение 1 — 2 мин.
Для анализа берут 2 мл прозрачного раствора. В испытуемый раствор вносят 1 мл раствора ванадий-оксихинолинового комплекса в бензоле. Перемешивают и нагревают на водяной бане при 65 — 70 °C в течение 30 мин. После охлаждения добавляют 1 мл раствора едкого натра. Пробирку вновь интенсивно встряхивают до исчезновения черной окраски. Фотометрируют при длине волны 370 нм в кювете с толщиной слоя 1 см по сравнению с контролем. Содержание спиртов в анализируемом объеме определяют по калибровочному графику.
Для построения калибровочного графика готовят шкалу стандартов. Для этого рабочий стандартный расчет наносят на уголь, употребляемый для отбора проб, одновременно готовят контроль (табл. 59).
Таблица 59
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
|
Рабочий стандартный раствор, мл |
0 |
0,05 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
|
Бензол, мл |
2 |
1,95 |
1,9 |
1,8 |
1,6 |
1,4 |
1,2 |
1 |
|
Содержание спирта, мкг |
0 |
5,0 |
10 |
20 |
40 |
60 |
80 |
100 |
Каждую пробирку шкалы и контроль обрабатывают аналогично пробе. Измеряют оптическую плотность и строят калибровочный график.
Шкала может быть использована для визуального определения. При этом ее готовят одновременно с пробами в колориметрических пробирках.
Расчет см. выше.
РАЗДЕЛЬНОЕ ОПРЕДЕЛЕНИЕ АЛИФАТИЧЕСКИХ СПИРТОВ
ГРУППЫ C1 — C10 ПРИ ИХ СОВМЕСТНОМ ПРИСУТСТВИИ С ПОМОЩЬЮ
БУМАЖНОЙ ХРОМАТОГРАФИИ [7]
Принцип определения
Спирты группы C1 — C10 переводят в соответствующие нелетучие производные 3,5-динитробензоата, которые разделяют затем на бумаге нисходящим методом в двух системах растворителей: диметилформамид-гексан (для C1 — C6) и вазелиновое масло — формамид (для C7 — C10).
После проявления хроматограммы производные спиртов обнаруживаются в виде желтых пятен. Количественное определение проводят фотометрически по изменению величины оптической плотности окрашенных элюатов, а также визуально сравнением интенсивности окраски проб с окраской «свидетелей».
Чувствительность по 0,5 мкг для каждого из спиртов C1 — C6 и 1 мкг для спиртов C6 — C10.
Определению не мешают алифатические кислоты группы C1 — C9, метиловые эфиры указанных кислот, амины C7 — C9, динил. Мешают определению соединения, содержащие гидроксильную группу.
Аппаратура
1. Теплоэлектровентилятор.
2. Пробирки с притертыми пробками емкостью 10 мл.
3. Хроматографические камеры размером 200 x 350 и 240 x 600 мм (рис. 26).
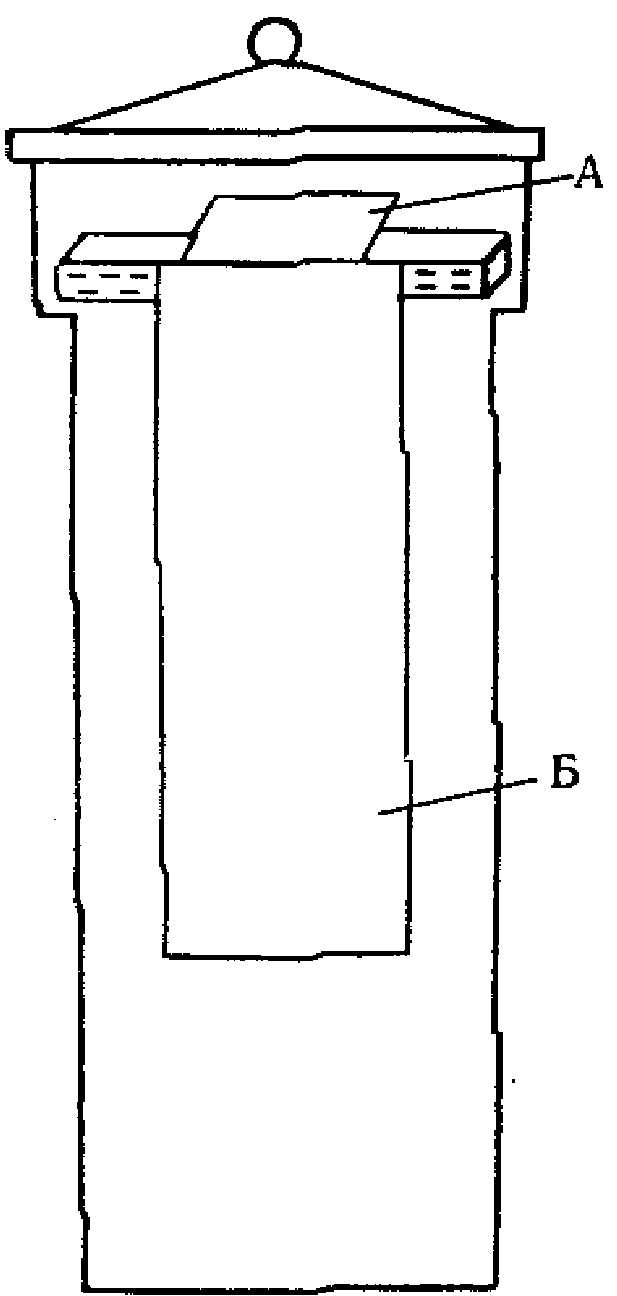
Рис. 26. Общий вид хроматографической камеры
А — лодочка с растворителем; Б — хроматографическая бумага
4. Делительные воронки емкостью 50 мл.
5. Эмалированная кювета размером 190 x 250 мм.
6. Мерные колбы емкостью 25 и 50 мл.
7. Микропипетки емкостью на 0,1 мл.
8. Резиновая груша.
9. Стеклянный распылитель.
10. Стеклянные трубки с четырьмя шариками для поглощения спиртов из воздуха (диаметр каждого шарика 10 мм).
11. Сушильный шкаф.
Реактивы
1. Активированный уголь марки АГ-5 обрабатывают концентрированной соляной кислотой в течение 1 — 1 1/2 ч при кипячении. Затем кислоту сливают, а уголь промывают несколько раз дистиллированной водой и 0,1 н. раствором аммиака до тех пор, пока последний не перестанет окрашиваться. Далее уголь снова промывают дистиллированной водой до отрицательной реакции на ион хлора и сушат тонким слоем при 100 — 120 °C в течение 1 — 2 ч. Хранят уголь в посуде с плотно прилегающей пробкой.
2. Стандартные растворы спиртов (метилового, этилового, пропилового, бутилового, амилового, гексилового, гептилового, октилового, нонилового, децилового) с содержанием 2 мг в 1 мл бензола.
3. 3,5-Динитробензоилхлорид — 10% раствор в бензоле (по весу).
4. Пиридин 10% раствор в бензоле (по объему).
5. Бензол.
6. Едкое кали 50% раствор.
7. Соляная кислота, 15% раствор.
8. Аммиак, 0,1% раствор.
9. Диметилформамид.
10. Гексан.
11. Вазелиновое масло.
12. Формамид.
13. Хлорид олова. Растворяют 0,7 г реагента в 5 мл воды, затем прибавляют 15 мл концентрированной соляной кислоты. Объем доводят до 100 мл водой. Раствор годен в течение суток.
14. П-Диметиламинобензальдегид. 1 г реагента растворяют в 95 мл этанола и прибавляют 5 мл концентрированной соляной кислоты. Раствор годен в течение 2 нед.
15. Метанол.
16. Ацетон.
17. Серная кислота, уд. вес 1,84.
18. Хроматографическая бумага марки «Ленинградская медленная» и «Ленинградская быстрая».
19. Этанол.
Отбор пробы
(см. выше «Определение общего количества алифатических спиртов» (C1 — C10).
Ход анализа
1. Получение 3,5-динитробензоатов спиртов
а) Обработка проб воздуха
После отбора пробы уголь переносят в пробирку с притертой пробкой и заливают 4 мл бензола. Содержимое пробирки встряхивают в течение 2 — 3 мин. и помещают пробирку на водяную баню, нагретую до 55 — 60 °C на один час. Затем пробирку вынимают из бани и снова встряхивают.
Для анализа берут 2 мл прозрачного раствора, переносят его в делительную воронку, куда добавляют 0,3 мл раствора 3,5-динитробензоилхлорида и 0,6 мл раствора пиридина. Объем пробы доводят до 5 мл бензолом, содержимое воронки встряхивают и оставляют на 30 мин. для образования 3,5-динитробензоатов спиртов.
Через 30 мин. производные спиртов промывают 5 мл раствора едкого кали, затем дважды водой по 25 мл каждый раз. Водный слой каждый раз удаляют, бензольный же промывают 5 мл 15% раствора соляной кислоты и дважды водой по 5 мл каждый раз.
Для получения более концентрированных проб бензол выпаривают на водяной бане. Полученный сухой остаток 3,5-динитробензоатов вновь растворяют в 0,15 мл бензола. 0,05 мл этого раствора используют для анализа спиртов группы C1 — C6 и 0,05 мл — для анализа спиртов C7 — C10.
б) Приготовление «свидетелей»
Стандартные растворы спиртов группы C1 — C6 смешивают в равных объемах по 0,5 мл, но не меньше, чем по 1 мг каждого. Так же поступают со стандартными растворами спиртов C7 — C10. Далее 3 мл смеси спиртов C1 — C6 (т.е. до 1 мг каждого спирта) помещают в делительную воронку. Дальнейшую обработку стандартных растворов спиртов проводят аналогично обработке проб воздуха.
Полученные растворы «свидетелей» содержат 3,5-динитробензоаты спиртов в количествах, соответствующих 0,4 мг/мл каждого из спиртов группы C1 — C10.
Производные спиртов могут сохраняться в течение 6 — 8 мес. при 6 — 8 °C.
2. Разделение 3,5-динитробензоатов
Для разделения производных спиртов C1 — C6 используется хроматографическая бумага марки «Ленинградская медленная». На листе размером 180 x 500 мм, отступя 7 см от края, проводят линию старта, на которой намечают четыре точки на расстоянии 3 — 3,5 см друг от друга.
С помощью микропипетки в одну точку наносят 0,05 мл раствора смеси соответствующих «свидетелей» спиртов группы C1 — C6, в три другие — пробы в количестве 0,05 мл. При этом диаметр пятен на линии старта не должен превышать 5 мм. С целью ускорения нанесения проб на бумагу можно использовать теплоэлектровентилятор. После нанесения пробы на бумагу последнюю «протягивают» через 50% раствор диметилформамида в ацетоне или этаноле, находящейся в эмалированной кювете. «Протягивают» бумагу от одного конца к другому до стартовой линии, не касаясь раствором проб, нанесенных на бумагу. Затем хроматографическую бумагу «отжимают» между листами фильтровальной бумаги и проделывают ту же операцию с другим ее концом. После этого бумагу подвешивают в течение 10 мин. для испарения растворителя, а затем помещают в хроматографическую камеру, предварительно насыщенную парами неподвижного и подвижного растворителей (диметилформамида и гексана).
Место соединения крышки с камерой заклеивают изоляционной лентой.
Разделение ведут нисходящим методом в течение 6 — 8 ч и считают законченным, когда подвижной растворитель — гексан — продвинется от линии старта на 40 см. После этого хроматограмму извлекают из камеры, сушат в течение 15 — 20 мин. на воздухе и проявляют.
Разделение производных спиртов групп C7 — C10 проводится на хроматографической бумаге марки «Ленинградская быстрая», которую после нанесения проб пропитывают 10% раствором вазелинового масла в гексане.
В качестве подвижной фазы используется формамид.
Разделение осуществляется в хроматографической камере размером 200 x 350 мм (см. рис. 26) и считается законченным, когда подвижной растворитель продвинется по бумаге на 25 см от линии старта. Далее хроматограмму сушат в вентилируемом сушильном шкафу при температуре 100 — 120 °C в течение 20 — 30 мин., а затем проявляют.
3. Обнаружение производных спиртов
Хроматограммы орошают раствором хлористого олова и через 1 — 2 ч раствором П-диметиламинобензальдегида. Производные спиртов C1 — C10 тотчас проявляются в виде круглых желтых пятен, окраска которых сохраняется более года.
Путем сравнения расположения на хроматограмме пятен проб с пятнами «свидетелей» делают выводы о наличии в пробах тех или иных спиртов, так как производные одного и того же спирта должны находиться на одинаковом расстоянии от линии старта.
Качественный состав пробы можно установить по величинам, которые вычисляют для каждого спирта отдельно (Rf — отношение расстояния пятна до линии старта к расстоянию от линии старта до линии фронта растворителя).
Затем, пользуясь табл. 60, находят отдельные спирты на хроматограмме.
Таблица 60
СРЕДНИЕ ВЕЛИЧИНЫ Rf ДЛЯ СПИРТОВ
|
Спирт |
Величина Rf в системе растворителей диметилформамид-гексан |
Спирт |
Величина Rf в системе растворителей вазелиновое масло-формамид |
|
CH3OH |
0,11 |
C7H13OH |
0,54 |
|
C2H5OH |
0,2 |
C8H17OH |
0,40 |
|
C3H7OH |
0,3 |
C9H19OH |
0,22 |
|
C4H9OH |
0,38 |
C10H21OH |
0,13 |
|
C5H11OH |
0,45 |
||
|
C6H13OH |
0,51 |
4. Количественное определение спиртов группы
C1 — C10 методом визуальной оценки
Готовят растворы «свидетелей» с различным содержанием производных спиртов. Для этого используют «свидетели», приготовленные согласно описанному выше способу. Бензол выпаривают, а сухие остатки, представляющие собой 3,5-динитробензоаты спиртов групп C1 — C6 и C7 — C10, растворяют каждый отдельно в 2,5 мл бензола (исходные растворы 1 и 2 соответственно).
Из полученных исходных растворов готовят серии разведений как показано в табл. 61 и 62.
Таблица 61
ПРИГОТОВЛЕНИЕ СЕРИИ «СВИДЕТЕЛЕЙ» ДЛЯ ОПРЕДЕЛЕНИЯ
СПИРТОВ ГРУПП C1 — C6
|
Исходный раствор спирта в бензоле, мл |
Бензол, мл |
Соответствующее количество каждого спирта, мкг |
|
|
в 1 мл |
в 0,05 мл |
||
|
2,5 |
0 |
400 |
20 |
|
0,8 |
0,2 |
320 |
16 |
|
0,6 |
0,4 |
240 |
12 |
|
0,4 |
0,6 |
160 |
8 |
|
0,2 |
0,8 |
80 |
4 |
|
0,05 |
0,95 |
20 |
1 |
Таблица 62
ПРИГОТОВЛЕНИЕ СЕРИИ СВИДЕТЕЛЕЙ ДЛЯ ОПРЕДЕЛЕНИЯ СПИРТОВ
ГРУППЫ C7 — C10
|
Исходный раствор спирта в бензоле, мл |
Бензол, мл |
Соответствующее количество каждого спирта, мкг |
|
|
в 1 мл |
в 0,05 мл |
||
|
2,5 |
0 |
400 |
20 |
|
0,6 |
0,4 |
240 |
12 |
|
0,4 |
0,6 |
160 |
8 |
|
0,2 |
0,8 |
80 |
4 |
|
0,05 |
0,95 |
20 |
1 |
Для определения спиртов на лист хроматографической бумаги в разные точки наносят по 0,05 мл раствора «свидетелей». Затем «свидетели» подвергаются хроматографическому анализу в тех же условиях, что и пробы.
На проявленных хроматограммах обнаруживаются пятна, разные по величине и интенсивности окраски и соответствующие 1 — 4 — 8 — 12 — 16 — 20 мкг каждого из спиртов групп C1 — C6 и 1 — 4 — 8 — 12 мкг — спиртов групп C7 — C10.
Количественное определение спиртов (после разделения их производных и проявления хроматограмм) методом визуальной оценки основано на том, что размер пятен и интенсивность окраски производных пропорциональны содержанию спиртов. Сравнивая интенсивность окраски пятен проб с окраской свидетелей, определяют содержание каждого спирта в пробе.
5. Количественное определение спиртов
групп C1 — C6 по оптической плотности элюатов
При фотометрическом определении содержания спиртов в пробе предварительно готовят стандартную шкалу, как описано выше, т.е. подвергают хроматографическому разделению «свидетели», соответствующие 1 — 4 — 8 — 12 — 16 — 20 мкг каждого из спиртов групп C1 — C6.
После проявления хроматограмм вырезают каждое пятно из бумаги, измельчают, помещают в пробирку и заливают 5 мл абсолютного метанола. Содержимое пробирки слегка встряхивают. Через 5 мин. окрашенный элюат переносят в кювету и измеряют величину его оптической плотности при длине волны 445 нм с помощью спектрофотометра или электрофотоколориметра.
Опыты повторяют 4 — 5 раз и по полученным средним данным строят градуировочные графики зависимости оптической плотности элюатов от концентрации вещества.
Такие графики строят для каждого спирта — метилового, этилового, пропилового, бутилового, амилового, гексилового.
Для определения содержания спиртов в пробах после их хроматографического анализа окраску каждого пятна элюируют, как описано выше, измеряют величину оптической плотности элюатов и с помощью графиков находят количество спиртов в пробе.
Расчет см. выше.
ИЗОПРОПИЛОВЫЙ СПИРТ (ИЗОПРОПАНОЛ)
Прозрачная бесцветная жидкость, температура кипения 82,8 °C, плотность при 20 °C 0,785, хорошо растворим в воде.
Общее действие на организм сходно с этанолом, но при равных концентрациях паров сильнее его.
Пары изопропанола раздражают слизистые оболочки глаз и верхних дыхательных путей, могут повредить сетчатку глаз и зрительный нерв.
Предельно допустимая концентрация: максимальная разовая 0,6 мг/м3.
ОПРЕДЕЛЕНИЕ ИЗОПРОПИЛОВОГО СПИРТА
С САЛИЦИЛОВЫМ АЛЬДЕГИДОМ [9]
Принцип определения
При взаимодействии изопропанола с персульфатом калия образуется ацетон, который с салициловым альдегидом в щелочной среде дает соединение, окрашивающее раствор от желтого до ярко-оранжевого цвета.
Чувствительность определения 2 мкг в анализируемом объеме раствора.
Ацетон мешает определению. Пропиловый спирт определению не мешает.
Реактивы
1. Поглотительный раствор — вода (дистиллированная).
2. Стандартный раствор. Готовят исходный раствор в мерной колбе емкостью 50 мл, наливают 5 мл воды и взвешивают. Затем в нее вносят 0,1 — 0,2 мл изопропанола и снова взвешивают. Раствор в колбе доводят до метки водой. По разности двух взвешиваний устанавливают содержание изопропанола в колбе и рассчитывают количество его в 1 мл раствора. Рабочий стандартный раствор с содержанием 10 мкг/мл получают в мерной колбе емкостью 50 мл, в нее вносят такое количество исходного раствора, в котором содержатся 500 мкг изопропанола. Раствор доводят до метки водой.
3. Изопропанол, температура кипения 82,8 °C.
4. Персульфат калия, 1% раствор.
5. Натр едкий, 40% раствор.
6. Альдегид салициловый, 20% раствор в этаноле.
7. Бисульфит калия, 5% раствор.
Отбор пробы
Для определения разовой концентрации воздух протягивают со скоростью 0,3 л/мин. через два поглотительных прибора с пористым фильтром N 1, наполненные по 2 мл воды каждый. Продолжительность отбора 20 — 30 мин.
Ход анализа
Каждый поглотительный прибор анализируют отдельно. В колориметрическую пробирку с пробкой переливают пробу (2 мл) и вносят в нее 0,2 мл персульфата калия. Пробирку быстро закрывают во избежание потери образовавшегося ацетона, ставят на водяную баню, которую постепенно нагревают до 50 — 54°, и при этой температуре оставляют пробирку на 30 мин. Затем пробирку вынимают и дают раствору охладиться при комнатной температуре. По охлаждении вносят 0,2 мл 5% раствора бисульфита и 2 мл 40% щелочи, содержимое пробирки встряхивают и вливают 0,2 мл салицилового альдегида. Раствор встряхивают и пробирку помещают в водяную баню, нагретую до 70 °C на 15 — 20 мин. По охлаждении пробу фотометрируют в кюветах с толщиной слоя 1 см при длине волны 445 — 450 нм по сравнению с контролем. Содержание изопропилового спирта в анализируемом объеме определяют по калибровочному графику. Для построения калибровочного графика готовят шкалу стандартов (табл. 63). Шкалу стандартов можно использовать для визуального определения.
Таблица 63
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
|
Рабочий стандартный раствор, мл |
0 |
0,2 |
0,4 |
0,8 |
1 |
1,5 |
2 |
|
Вода, мл |
2 |
1,8 |
1,6 |
1,2 |
1,0 |
0,5 |
0 |
|
Содержание изопропанола, мкг |
0 |
2 |
4 |
8 |
10 |
15 |
20 |
Все пробирки шкалы обрабатывают аналогично пробе.
Расчет см. выше.
ЦИКЛОГЕКСАНОЛ
Бесцветная жидкость, обладающая запахом камфоры, температура кипения 161 °C (технического 155 — 165), плотность 0,962 при 20 °C, растворима в воде.
Циклогексанол раздражающе действует на слизистую оболочку глаз.
Предельно допустимые концентрации: максимальная разовая и среднесуточная 0,06 мг/м3.
ОПРЕДЕЛЕНИЕ ЦИКЛОГЕКСАНОЛА
С ПАРАДИМЕТИЛАМИНОБЕНЗАЛЬДЕГИДОМ [6]
Принцип определения
При взаимодействии циклогексанола с парадиметиламинобензальдегидом в сернокислой среде образуется соединение, окрашивающее раствор в розовый цвет.
Чувствительность определения 0,8 мкг в анализируемом объеме раствора.
Амины, высшие спирты мешают определению.
Реактивы
1. Поглотительный раствор — дистиллированная вода.
2. Стандартный раствор циклогексанола: готовят исходный раствор в мерной колбе емкостью 50 мл, в которую наливают 5 — 10 мл воды, и взвешивают. В колбу вносят 2 — 3 капли циклогексанола и снова взвешивают. По разности двух взвешиваний устанавливают количество циклогексанола. Объем жидкости в колбе доводят до метки водой. Вычисляют количество циклогексанола в 1 мл.
Рабочий стандартный раствор с содержанием 10 мкг/мл готовят в мерной колбе емкостью 50 мл, в нее вносят такое количество исходного раствора, в котором содержится 500 мкг циклогексанола. Раствор доводят до метки водой.
3. Серная кислота, уд. вес 1,84 и 1,727, т.е. 80%.
4. Парадиметиламинобензальдегид, 0,5% раствор в 80% серной кислоте.
Отбор пробы
Для определения разовой концентрации воздух протягивают со скоростью 0,5 л/мин. через поглотительный прибор с пористым фильтром N 1, содержащий 3 мл дистиллированной воды. Продолжительность отбора 20 — 30 мин.
Ход анализа
В пробирку помещают 2 мл пробы, в которую наливают 0,2 мл парадиметиламинобензальдегида и затем 4 мл серной кислоты удельного веса 1,84. Содержимое пробирки встряхивают и помещают на 10 мин. в кипящую водяную баню. По охлаждении раствора измеряют оптическую плотность в кювете толщиной слоя 1 см по сравнению с контролем, который готовят одновременно и аналогично пробе. Определение проводят при длине волны 510 нм.
Содержание циклогексанола в анализируемом объеме определяют по предварительно построенному калибровочному графику.
Для построения графика готовят шкалу стандартов (табл. 64).
Таблица 64
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
|
Рабочий стандартный раствор, мл |
0 |
0,08 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
|
Вода, мл |
2 |
1,92 |
1,9 |
1,8 |
1,6 |
1,4 |
1,2 |
1,0 |
|
Содержание циклогексанола, мкг |
0 |
0,8 |
1 |
2 |
4 |
6 |
8 |
10 |
Все пробирки шкалы обрабатывают аналогично пробе.
Шкалу стандартов можно использовать для визуального определения, шкалу готовят в колориметрических пробирках одновременно с пробой.
Расчет см. выше.
ФЕНОЛ
(Оксибензол, карболовая кислота)
Бесцветные кристаллы, краснеющие на воздухе.
Температура плавления 41 °C, температура кипения 182 °C, растворяется в воде 6,7 г в 100 мл при температуре 16 °C, при 66 °C смешивается с водой во всех отношениях.
Растворим в этаноле, эфире, хлороформе и других органических растворителях.
При вдыхании паров фенола наблюдаются раздражение дыхательных путей, расстройство пищеварения, общая и мышечная слабость и другие патологические явления.
При попадании фенола на кожу в местах соприкосновения с фенолом происходит омертвение тканей, общее отравление организма, нарушаются функции кровообращения, дыхания, повышается температура.
Предельно допустимые концентрации: максимальная разовая 0,01 мг/м3, среднесуточная 0,01 мг/м3.
ОПРЕДЕЛЕНИЕ ФЕНОЛА С П-НИТРОФЕНИЛДИАЗОНИЕМ [4]
Принцип определения
При взаимодействии фенола с п-нитрофенилдиазонием в щелочной среде образуется продукт, окрашенный в красный цвет.
Чувствительность определения 0,5 мкг в определяемом объеме раствора.
Определению мешают орто-, метакрезолы, амины.
Реактивы
1. Поглотительный раствор — 0,8% раствор карбоната натрия.
2. Силикагель КСМ, КСК, величина зерен 0,5 — 2 мм (предварительно обработанный) (см. ниже).
3. Стандартный раствор фенола. Исходный раствор с содержанием (приблизительно) 0,5 мг фенола в 1 мл готовят растворением около 0,125 г фенола в мерной колбе емкостью 250 мл. Доводят до метки 0,8% раствором карбоната натрия. Точное содержание вычисляют по навеске.
Рабочий стандартный раствор с содержанием 10 мкг в 1 мл готовят соответствующим разбавлением исходного.
Рабочие растворы используют свежеприготовленными.
4. Нитрит натрия, 25% раствор (свежеприготовленный).
5. Паранитроанилин.
6. Соляная кислота, уд. вес 1,19.
7. П-Нитрофенилдиазоний. Готовят добавлением к 50 мл воды 2,5 мл соляной кислоты уд. веса 1,19, 2,5 мл 25% раствора нитрита натрия и 0,01 г паранитроанилина. Тщательно перемешивают. Раствор применяется свежеприготовленным.
8. Этанол.
Отбор пробы
а) Для определения разовой концентрации отбирают пробу воздуха в «кипящий» слой силикагеля. Для этого по 3 мл силикагеля вносят через воронку в два поглотительных прибора НИИ гигиены имени Ф.Ф. Эрисмана, соединенные последовательно, к которым присоединен поглотитель для улавливания переброшенных частиц силикагеля. Воздух протягивают со скоростью до 6 л/мин. Продолжительность отбора 20 — 30 мин.
б) Для определения среднесуточной концентрации воздух со скоростью 2 — 3 л/мин. протягивают в «кипящий» слой силикагеля в течение суток непрерывно.
Допустимо проводить отбор в одну и ту же систему поглотителей 6 — 12 раз с перерывом в 2 — 4 ч.
При определении концентраций фенола, превышающих допустимые, воздух со скоростью 0,5 л/мин. протягивают через два последовательно соединенных поглотительных прибора с пористым фильтром N 1, наполненных по 6 мл поглотительного раствора.
Ход анализа
а) При отборе проб на силикагель последний из каждого поглотительного прибора отдельно переносят в пробирки с притертыми пробками. Переброшенные зерна силикагеля (из последнего прибора) присоединяют к силикагелю из второго поглотительного прибора.
Добавляют по 2 мл этанола, перемешивают и через 30 мин. добавляют по 2 мл поглотительного раствора. Вновь взбалтывают и дают отстояться. Для анализа берут по 2 мл прозрачного раствора.
Одновременно готовят шкалу стандартов и контроль. Для этого в пробирки помещают по 3 мл силикагеля, употребляемого при отборе, наносят на силикагель стандартные растворы фенола и другие реактивы, согласно табл. 65.
Таблица 65
ШКАЛА СТАНДАРТОВ ПРИ ОТБОРЕ ПРОБ НА СИЛИКАГЕЛЬ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
|
Рабочий стандартный раствор, мл |
0 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
|
Этанол, мл |
По 1 мл во все пробирки |
||||||
|
Поглотительный раствор, мл |
1 |
0,9 |
0,8 |
0,6 |
0,4 |
0,2 |
0,0 |
|
Содержание фенола, мкг |
0 |
1,0 |
2,0 |
4,0 |
6,0 |
8,0 |
10,0 |
Стандартную шкалу обрабатывают аналогично пробам.
Содержание фенола устанавливают путем визуального сравнения интенсивности окраски пробы со шкалой.
б) При отборе проб в жидкую поглотительную среду содержимое каждого поглотительного прибора переносят в пробирки. Для анализа берут 5 мл пробы. Добавляют по 0,2 мл раствора п-нитрофенилдиазония. Содержимое пробирки встряхивают и через 10 мин. фотометрируют при длине волны 495 нм в кювете с толщиной слоя 1 см по сравнению с контролем.
Содержание фенола в анализируемом объеме определяют по калибровочному графику. Для построения калибровочного графика готовят шкалу стандартов (табл. 66).
Таблица 66
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
|
Рабочий стандартный раствор, мл |
0 |
0,05 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
1,5 |
|
Поглотительный раствор, мл |
5 |
4,95 |
4,9 |
4,8 |
4,6 |
4,4 |
4,2 |
4,0 |
3,5 |
|
Содержание фенола, мкг |
0 |
0,5 |
1,0 |
2,0 |
4,0 |
6,0 |
8,0 |
10,0 |
15,0 |
В каждую пробирку шкалы стандартов добавляют по 0,2 мл раствора п-нитрофенилдиазония, перемешивают, измеряют оптическую плотность и строят калибровочный график.
Шкала стандартов может быть использована для визуального определения. В этом случае ее готовят в колориметрических пробирках одновременно с пробами.
ОПРЕДЕЛЕНИЕ ФЕНОЛА С 4-АМИНОАНТИПИРИНОМ [10]
Принцип определения
При взаимодействии фенола с 4-аминоантипирином в присутствии железосинеродистого калия при pH среды 9,3 образуется продукт, окрашенный в красный цвет.
Чувствительность — 0,5 мкг в определяемом объеме раствора.
Мешает определению ацетофенон в количествах, превышающих 20 мкг. Не мешают определению паракрезол, бензол, изопропилбензол, гидроперекись изопропилбензола, ацетон, стирол, ![]() -метилстирол, диметилвинилпараоксифенилметан.
-метилстирол, диметилвинилпараоксифенилметан.
Реактивы
1. Поглотительный раствор — тетраборат натрия Na2B4O7 ·10H2O, 0,05 М раствор. Готовят растворением 1 г тетрабората в 1 л воды.
2. Силикагель КСК, КСМ, величина зерен 0,5 — 2 мм (предварительно обработанный, см. ниже).
3. Стандартные растворы фенола с содержанием 500; 10 и 1 мкг/мл готовят так же, как указано в методе с п-нитрофенилдиазонием.
4. 4-Аминоантипирин, 1% раствор.
5. Железосинеродистый калий (K3[Fe(CN)6]), 0,1% раствор.
6. Этанол.
Отбор проб
См. «Определение фенола с п-нитрофенилдиазонием».
Ход анализа
а) При отборе проб в жидкую среду содержимое каждого поглотительного прибора переносят в пробирки. Для анализа берут 5 мл пробы. Добавляют по 0,1 мл 1% раствора 4-аминоантипирина, по 0,1 мл 0,1% раствора железосинеродистого калия.
Содержимое пробирок перемешивают и через 10 мин. фотометрируют при длине волны 495 нм в кювете с толщиной слоя 1 см по сравнению с контролем. Содержание фенола в анализируемом объеме определяют по калибровочному графику. Для построения калибровочного графика готовят шкалу стандартов (табл. 67).
Таблица 67
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
|
Рабочий стандартный раствор, мл |
0 |
0,05 |
0,1 |
0,2 |
0,3 |
0,4 |
0,5 |
0,6 |
|
Поглотительный раствор, мл |
5 |
4,95 |
4,9 |
4,8 |
4,7 |
4,6 |
4,5 |
4,4 |
|
Содержание фенола, мкг |
0 |
0,5 |
1,0 |
2,0 |
3,0 |
4,0 |
5,0 |
6,0 |
В каждую пробирку шкалы вносят по 0,1 мл 1% раствора 4-аминоантипирина и поступают аналогично анализу пробы.
Шкала стандартов может быть использована для визуального определения. Ее в этом случае готовят одновременно с пробами в колориметрических пробирках.
б) При отборе проб на силикагель поступают так же, как и при определении фенола с п-нитрофенилдиазонием.
После добавления к силикагелю этилового спирта вносят в пробу по 2 мл раствора тетрабората натрия и далее все реактивы в том же порядке, как при определении фенола, поглощенного в жидкую среду.
Интенсивность окраски проб визуально сравнивают с интенсивностью окраски шкалы (табл. 67).
Расчет см. выше.
СЛОЖНЫЕ ЭФИРЫ
RCOOR1
Сложные эфиры уксусной кислоты — бесцветные летучие жидкости, обладающие характерным запахом. Растворимость их в воде ограничена и уменьшается с увеличением молекулярного веса. Легко воспламеняются, в определенной смеси с воздухом взрывоопасны. Относятся к токсическим веществам, их пары раздражают слизистые оболочки дыхательных путей, глаз.
Таблица 68
НЕКОТОРЫЕ ФИЗИЧЕСКИЕ СВОЙСТВА И ПРЕДЕЛЬНО ДОПУСТИМЫЕ
КОНЦЕНТРАЦИИ (ПДК) ЭФИРОВ УКСУСНОЙ КИСЛОТЫ
|
Название |
Формула |
Молекулярный вес |
Плотность |
Температура кипения при 760 мм рт. ст. |
ПДК мг/м3 |
|
|
максимальная разовая |
среднесуточная |
|||||
|
Амилацетат |
CH3COOC2H5 |
130,19 |
0,879 (20°) |
142,5 |
0,1 |
0,1 |
|
Бутилацетат |
CH3COOC4H9 |
116,16 |
0,883 (20°) |
125 (740 мм рт. ст.) |
0,1 |
0,1 |
|
Винилацетат |
CH3COOCH2CH2 |
86,09 |
0,932 (20°) |
72,3 |
0,3 |
0,2 |
|
Метилацетат |
CH3COOCH3 |
74,08 |
0,934 |
57,3 |
0,07 |
0,07 |
|
Этилацетат |
CH3COOC2H5 |
88,10 |
0,900 |
77,15 |
0,1 |
0,1 |
ОПРЕДЕЛЕНИЕ СЛОЖНЫХ ЭФИРОВ (АМИЛАЦЕТАТА, БУТИЛАЦЕТАТА,
ВИНИЛАЦЕТАТА, МЕТИЛАЦЕТАТА, ЭТИЛАЦЕТАТА)
С СОЛЯНОКИСЛЫМ ГИДРОКСИЛАМИНОМ [5]
Принцип определения
При взаимодействии сложных эфиров с солянокислым гидроксиламином в щелочной среде образуются гидроксаматы, которые реагируют с хлорным железом, давая продукт, окрашенный в фиолетовый цвет.
Чувствительность определения 5 мкг в анализируемом объеме раствора.
Ангидриды кислот, сложные эфиры адипиновой, акриловой, малеиновой кислот мешают определению.
Реактивы
1. Силикагель АСМ, КСМ, КСК с величиной зерен 0,25 — 1 мм, обработанный соляной кислотой, разбавленной водой в отношении 1:1 при нагревании в течение 3 — 4 ч, промывают водой. После этого силикагель высушивают при 100 — 105 °C и активируют при 200 °C в течение 30 мин.
Регенерация силикагеля проводится тем же способом.
2. Этанол, предварительно обработанный едкой щелочью. Для этого в колбу емкостью 500 мл всыпают 20 — 30 г едкого натра, вливают этанол, раствор встряхивают и оставляют на 2 — 3 ч, временами встряхивая его. После этого раствор перегоняют при 78 °C.
3. Стандартные растворы сложных эфиров готовят в этаноле. В мерную колбу емкостью 25 — 30 мл вливают 10 — 15 мл этанола, взвешивают, вносят в колбу эфир, который предполагают определять, 4 — 5 капель, и снова взвешивают. По разности двух взвешиваний определяют количество эфира и вычисляют его содержание в 1 мл раствора. Объем раствора доводят до метки этанолом. Рабочий стандартный раствор с содержанием 100 мкг/мл готовят в мерной колбе емкостью 50 мл, в которую вливают количество исходного раствора с содержанием 5000 мкг эфира. Объем раствора доводят до метки этанолом.
4. Гидроксиламин солянокислый, 4 н. раствор.
5. Едкий натр, 4 н. раствор.
6. Соляная кислота, 0,5 н. и 4 н. раствор.
7. Хлорное железо окисное, 1,6% раствор в 0,5 н. растворе соляной кислоты.
8. Фенолфталеин, 0,5% спиртовой раствор.
Отбор пробы
а) Для определения разовой концентрации воздух со скоростью 5 — 10 л/мин. протягивают в «кипящий» слой силикагеля. Для этого в два последовательно присоединенных поглотительных прибора НИИ гигиены имени Ф.Ф. Эрисмана или поглотителя Яворовской помещают по 3 мл силикагеля, к ним присоединяют прибор для улавливания переброшенных частиц силикагеля.
Винилацетат поглощают на неподвижный слой силикагеля, помещенный в шариковую трубку.
Продолжительность отбора 20 — 30 мин.
б) При определении среднесуточной концентрации отбор проб проводят в «кипящий» слой силикагеля со скоростью 1 — 2 л/мин. в течение суток непрерывно. Допустимо отбор проводить 6 — 12 раз с перерывами в 4 — 2 ч по 20 — 30 мин.
Ход анализа
Силикагель переносят из каждого поглотительного прибора отдельно в пробирки с притертыми пробками, добавляют по 6 мл этанола и помещают пробирки на водяную баню, нагретую до 50 °C на 20 мин. Содержимое пробирок периодически встряхивают. Раствору дают отстояться. Для определения берут по 3 мл прозрачного раствора, в пробирку вливают 0,2 мл 4 н. раствора гидроксиламина, 0,6 мл 4 н. раствора едкого натра. Раствор встряхивают и оставляют на 15 мин. Затем раствор нейтрализуют 4 н. соляной кислотой по фенолфталеину и в пробирку вливают 2,5 мл 1,6% раствора хлорида железа. Раствор встряхивают и через 20 — 30 мин. переливают в кювету толщиной слоя 1 см и измеряют оптическую плотность раствора, сравнивая с контролем, который готовят одновременно и аналогично пробе. Определение проводят с помощью фотоэлектроколориметра при длине волны 500 нм.
Содержание эфира в анализируемом объеме определяют по градуировочному графику, который предварительно строят по шкале стандартов (табл. 69).
Таблица 69
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
|
Рабочий стандартный раствор, мл |
0 |
0,05 |
0,10 |
0,20 |
0,30 |
0,40 |
0,5 |
|
Этанол, мл |
1,5 |
1,45 |
1,40 |
1,30 |
1,20 |
1,10 |
1,0 |
|
Содержание эфира, мкг |
0 |
5 |
10 |
20 |
30 |
40 |
50 |
Все пробирки шкалы обрабатывают аналогично пробе.
Шкалу стандартов можно использовать для визуального определения, готовят ее одновременно с пробами в колориметрических пробирках. Первая пробирка содержит 2 мкг эфира.
Расчет см. выше.
Примечание. При концентрации эфиров выше допустимой нормы отбор проб можно проводить в спирт со скоростью 0,3 — 0,5 л/мин.
ОПРЕДЕЛЕНИЕ ВИНИЛАЦЕТАТА ХРОМАТОГРАФИЕЙ НА БУМАГЕ [11]
Принцип определения
При взаимодействии винилацетата с ацетатом ртути образуется нелетучее ртутноорганическое соединение. Его выделяют на бумаге в системе растворителей бутанол — триэтиламин — вода нисходящим способом. Проявляющим реактивом служит раствор дифенилкарбазида.
Чувствительность 3 мкг в пробе. Определению не мешают 2-этилгексилакрилат, винилхлорид, дибутилмалеинат, диоктилфталат, дибутилфталат, стирол, ![]() -метилстирол.
-метилстирол.
Аппаратура
1. Электровентилятор.
2. Хроматографическая камера размером 260 x 600 мм с лодочкой и подставкой для лодочки (см. рис. 26).
3. Микропипетки емкостью 0,1 мл с делениями на 0,01 мл.
4. Стеклянный распылитель (пульверизатор).
Реактивы
1. Винилацетат, перегнанный при 72,5 °C. Исходный стандартный раствор готовят в мерной колбе емкостью 10 мл, в которую вносят 2 — 3 мл поглотительного раствора, колбу взвешивают и добавляют 1 — 2 капли винилацетата. Раствор в колбе доводят до метки поглотительным раствором. Рассчитывают содержание винилацетата в 1 мл раствора. Соответствующим разбавлением готовят раствор N 1 с содержанием 200 мкг/мл. Рабочие стандартные растворы готовят из раствора N 1 в соответствии с табл. 70.
Таблица 70
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
|
Стандартный раствор N 1, мл |
0 |
0,45 |
0,60 |
0,75 |
1,05 |
1,5 |
|
Поглотительный раствор, мл |
3 |
2,55 |
2,40 |
2,25 |
1,95 |
1,6 |
|
Содержание винилацетата в 1 мл раствора, мкг |
0 |
30 |
40 |
50 |
70 |
100 |
2. Поглотительный раствор: к 100 мл пропилового спирта добавляют 0,5 мл ледяной уксусной кислоты и 0,2 г ацетата ртути.
3. Пропиловый спирт.
4. Бутиловый спирт.
5. Ацетат ртути, двузамещенная соль.
6. Кислота уксусная, ледяная.
7. Триэтиламин.
8. Дифенилкарбазид, 0,15% раствор.
9. Этиловый спирт очищенный. К 1 л спирта добавляют 40 г едкого кали, оставляют на 4 ч, затем отгоняют при 78,8 °C.
10. Система растворителей. В делительную воронку вносят 25 мл бутанола, 7 мл триэтиламина и 27 мл воды. Смесь встряхивают в течение 10 мин. и оставляют до полного расслоения.
11. Бумага хроматографическая «Медленная».
Отбор пробы
Воздух со скоростью 1 л/мин. протягивают через две пары параллельно соединенных поглотительных приборов с пористой пластинкой N 2, содержащих по 3 мл поглотительного раствора.
Отбор проводят в течение 20 — 30 мин. при охлаждении.
Ход анализа
Жидкость из первых двух поглотительных приборов переносят в центрифужные пробирки с делениями. Также объединяют и поглотительные жидкости из вторых поглотительных приборов.
Пробирки помещают в кипящую водяную баню и выпаривают спирт до объема 0,3 мл (по метке).
В хроматографической камере по стенкам располагают фильтровальную бумагу и смачивают ее нижним слоем, полученным при разделении слоев жидкостей системы, предназначенной для разделения смеси. Верхний слой жидкости переносят из делительной воронки в лодочку. Насыщение хроматографической камеры парами растворителей проводят в течение 4 — 5 ч.
На полоске хроматографической бумаги на расстоянии 6 см от края намечают линию старта, на которой отмечают три точки на расстоянии 2,5 см друг от друга, в которые при помощи микропипетки дробно наносят пробу, обдувая теплым воздухом. Затем пробирку обмывают 0,1 — 0,15 мл пропилового спирта и смыв наносят в те же точки. Хроматографическую бумагу с нанесенной пробой помещают в лодочку, находящуюся в хроматографической камере. Камеру герметизируют и оставляют на 15 — 17 ч при комнатной температуре. При этом растворитель опускается по бумаге на расстояние около 25 см.
Далее бумагу извлекают из камеры, высушивают при комнатной температуре, орошают раствором дифенилкарбазида и термостатируют в течение 3 мин. при 60 — 70 °C. При наличии винилацетата на хроматограмме проявляются зоны локализации в виде круглых пятен, окрашенных в фиолетовый цвет, с величиной Rf 0,7. На линии старта также проявляются окрашенные зоны, свидетельствующие об избытке ацетата ртути.
Зоны, характерные для винилацетата, вырезают из бумаги, измельчают и помещают в пробирку, в которую вносят 4 мл этилового спирта. Содержимое пробирки встряхивают трижды в течение 15 — 20 мин., после чего раствор центрифугируют.
Оптическую плотность измеряют на спектрофотометре в кюветах с толщиной слоя 1 см при длине волны 560 нм.
Содержание винилацетата устанавливают по калибровочному графику. Для построения калибровочного графика готовят несколько стандартных шкал.
Согласно шкалы стандартов (табл. 70), получают ряд растворов — N 1 — 2 — 3 и т.д. с содержанием от 30 до 100 мкг винилацетата в 1 мл.
На другой лист хроматографической бумаги по линии старта в отдельные точки наносят по 0,1 мл указанных в табл. 70 растворов, что соответствует N 3 — 4 — 5 — 7 и 10 мкг винилацетата.
Одновременно готовят контроль. При этом в контрольную точку наносят 0,1 мл поглотительного раствора. Бумагу с нанесенной стандартной шкалой помещают в ту же лодочку, в которую помещена проба, располагая ее по другую сторону хроматографической камеры. При этом достигают идентичности условий хроматографирования.
После хроматографирования шкалу орошают раствором дифенилкарбазида, производят те же операции и измеряют величины оптических плотностей, после чего строят калибровочный график.
Шкалой стандартов можно пользоваться для визуального определения.
Расчет см. выше.
МЕТИЛОВЫЙ ЭФИР МЕТАКРИЛОВОЙ КИСЛОТЫ
(метилметакрилат)
|
CH2=C(CH3)COOCH3 |
Мол. вес 100,11 |
Прозрачная жидкость, температура кипения 100 °C, плотность при 15,6 °C 0,95, нерастворим в воде, хорошо растворим в органических растворителях. Легко полимеризуется; гидрохинон, пирогаллол задерживают полимеризацию.
Обладает наркотическим и общетоксическим действием.
Предельно допустимые концентрации: максимальная разовая и среднесуточная 0,1 мг/м3.
ОПРЕДЕЛЕНИЕ МЕТИЛМЕТАКРИЛАТА ПО ФОРМАЛЬДЕГИДУ [1]
Принцип определения
При омылении метилметакрилата щелочью образуется метанол, который окисляют до формальдегида. Последний определяют с хромотроповой кислотой.
Чувствительность определения — 2 мкг в анализируемом объеме раствора.
Метанол, формальдегид, метиловые эфиры других кислот мешают определению.
Реактивы
1. Поглотительный раствор — 2,5% раствор едкого натра.
2. Стандартный раствор. Готовят исходный раствор в мерной колбе емкостью 50 мл, вливают 5 — 10 мл 25% раствора едкого натра, колбу взвешивают и вносят 2 — 3 капли метилметакрилата, снова взвешивают и объем в колбе доводят до метки 2,5% раствором едкого натра. По разности двух взвешиваний устанавливают количество метилметакрилата в колбе и вычисляют содержание его в 1 мл раствора.
Рабочий стандартный раствор с содержанием 10 мкг/мл получают в другой колбе емкостью 50 мл, в нее вносят такое количество исходного раствора, в котором содержится 500 мкг метилметакрилата. Объем раствора доводят до метки 2,5% раствором едкого натра.
3. Едкий натр, 2,5% раствор.
4. Серная кислота, разбавленная водой в отношении 1:1.
5. Перманганат калия, 2% раствор.
6. Сульфит натрия, 30% раствор, свежеприготовленный.
7. Хромотроповая кислота или ее динатриевая соль; 0,1 г этой кислоты растворяют в 2 мл воды и добавляют до 50 мл 15 М серной кислотой.
8. 15 М серная кислота. К 10 мл воды осторожно приливают при охлаждении 90 мл серной кислоты уд. веса 1,82 — 1,84.
9. Серная кислота, уд. вес 1,82 — 1,84.
Отбор пробы
а) Для определения разовой концентрации воздух протягивают со скоростью 0,5 л/мин. через два поглотительных прибора с пористым фильтром N 1, наполненных по 4 мл поглотительного раствора. Продолжительность отбора 20 — 30 мин.
б) Для определения среднесуточной концентрации непрерывно в течение суток воздух со скоростью 0,3 л/мин. протягивают через указанные выше приборы, содержащие по 4 мл поглотительного раствора. При наполнении прибора отмечают уровень жидкости, который периодически добавляют водой до первоначального.
Допустимо проводить отбор 6 — 12 раз с перерывом в 2 — 4 ч в одни и те же приборы по 20 — 30 мин.
Ход анализа
Анализируют каждый поглотительный прибор отдельно. Берут 2 мл пробы в пробирку, вливают 0,5 мл серной кислоты, разбавленной водой в отношении 1:1, и по капле раствора перманганата калия. Растворы взбалтывают и через 5 — 10 мин. вносят по одной капле сульфита для обесцвечивания раствора. Избытка сульфита необходимо избегать.
Затем вливают 2 мл хромотроповой кислоты. Раствор встряхивают и помещают на 5 мин. в кипящую водяную баню.
Раствор охлаждают и измеряют оптическую плотность раствора в кювете шириной 1 см по сравнению с контролем при длине волны 570 нм. Содержание метилметакрилата в анализируемом объеме определяют по предварительно построенному градуировочному графику.
Для построения графика готовят шкалу стандартов (табл. 71).
Таблица 71
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
|
Рабочий стандартный раствор, мл |
0 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
1,5 |
2,0 |
|
Поглотительный раствор, мл |
2 |
1,8 |
1,6 |
1,4 |
1,2 |
1,0 |
0,5 |
— |
|
Содержание метилметакрилата, мкг |
0 |
2 |
4 |
6 |
8 |
10 |
15 |
20 |
Все пробирки шкалы обрабатывают аналогично пробе. Шкалу стандартов можно использовать для визуального определения, ее следует готовить в колориметрических пробирках одновременно с пробой, шкалу начинать с 1 мкг и далее, как указано в табл. 71.
Расчет см. выше.
МЕТИЛОВЫЙ ЭФИР АКРИЛОВОЙ КИСЛОТЫ
(Метилакрилат)
|
CH2=CH-COO·CH3 |
Мол. вес 86,09 |
Прозрачная бесцветная жидкость с резким запахом, температура кипения 80 °C, плотность при 20 °C 0,95, незначительно растворим в воде, но хорошо растворим в органических растворителях, легко полимеризуется. Обладает наркотическим и общетоксическим действием на организм, а также резко раздражающим действием.
Предельно допустимые концентрации: максимальная разовая и среднесуточная — 0,04 мг/м3.
ОПРЕДЕЛЕНИЕ МЕТИЛАКРИЛАТА ПО ФОРМАЛЬДЕГИДУ [1]
Определение метилакрилата проводится так же, как метилметакрилата. Все реактивы те же, кроме стандартного раствора, его готовят из метилакрилата (см. выше).
ТЕТРАГИДРОФУРАН (ТГФ)
(Окись диэтилена)
|
|
Мол. вес 72,06 |
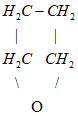
Жидкость с запахом этилового эфира, удельный вес ее при 20 °C 0,889, температура кипения 64 — 66 °C, смешивается с водой и спиртом в любых отношениях. Упругость пара ТГФ при 20 °C составляет 131,50, испаряется в 2,8 раза медленнее этилового эфира. ТГФ в промышленности применяется как растворитель и инсектицид. Тетрагидрофуран обладает наркотическим действием, раздражающим действием на слизистые оболочки глаз и верхние дыхательные пути, а также вызывает поражение почек.
Предельно допустимые концентрации ТГФ: максимальная разовая 0,2 мг/м3, среднесуточная 0,2 мг/м3.
ОПРЕДЕЛЕНИЕ ТЕТРАГИДРОФУРАНА
С ПАРАДИМЕТИЛАМИНОБЕНЗАЛЬДЕГИДОМ [3]
Принцип определения
При взаимодействии ТГФ с п-диметиламинобензальдегидом в сернокислой среде образуется окрашенный в розовый цвет продукт конденсации.
Чувствительность определения — 5 мкг ТГФ в пробе. Определению мешают фуран, спирты, а также соединения, дающие бурую окраску для реакции с серной кислотой. Фурфурол определению не мешает.
Реактивы
1. Силикагель марки АСМ или КСМ с зернами размером 0,3 — 0,5 мм.
Обработку силикагеля см. выше. Обработанный силикагель не должен давать реакцию на ТГФ.
2. Стандартные растворы ТГФ.
Исходный стандартный раствор готовят в мерной колбе емкостью 50 мл, куда вносят 10 мл воды, взвешивают и вносят несколько капель ТГФ, вновь взвешивают, доводят до метки и по разности весов устанавливают содержание ТГФ в 1 мл.
Соответствующим разбавлением готовят рабочий стандартный раствор с содержанием 100 мкг ТГФ в 1 мл.
3. 2% спиртовой раствор парадиметиламинобензальдегида.
4. Спирт, 96° ректификат.
5. Серная кислота концентрированная.
6. Соляная кислота, 1:2.
Отбор пробы
Для определения разовой концентрации в два последовательно соединенных поглотительных прибора НИИ гигиены имени Ф.Ф. Эрисмана засыпают по 3 мл силикагеля и протягивают анализируемый воздух со скоростью 5 — 10 л/мин. в кипящий слой силикагеля. Для улавливания переброшенных частиц силикагеля после второго поглотительного прибора ставят ловушку. Переброшенный силикагель присоединяют к силикагелю из второго поглотительного прибора. Отбор проводят в течение 20 — 30 мин.
Ход анализа
Силикагель переносят в две пробирки с притертыми пробками и заливают 2 мл дистиллированной воды и 8 мл концентрированной серной кислоты. Добавляют по 0,2 мл 2% раствора п-диметиламинобензальдегида. Содержимое пробирок осторожно встряхивают и выдерживают в кипящей водяной бане в течение часа.
В отдельной пробирке такой же обработке подвергается силикагель, не содержащий ТГФ (контроль).
По охлаждении пробирок окрашенный раствор над силикагелем сливают в кюветы с толщиной слоя 2 см и пробы фотометрируют при длине волны 520 нм.
Содержание ТГФ в пробе определяют по предварительно построенному калибровочному графику. Для построения калибровочного графика готовят шкалу стандартов (табл. 72), при этом стандартный раствор наносят на 3 мл силикагеля, помещенного в пробирки.
Таблица 72
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
|
Рабочий стандартный раствор, мл |
0 |
0,05 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
|
Вода, мл |
2 |
1,95 |
1,9 |
1,8 |
1,6 |
1,4 |
1,2 |
|
Серная кислота, мл |
По 8 мл |
||||||
|
Содержание ТГФ, мл |
0 |
5 |
10 |
20 |
40 |
60 |
80 |
Все пробирки шкалы обрабатывают аналогично пробам, измеряют оптическую плотность и строят график.
Шкалой стандартов можно пользоваться и для визуального определения, при этом растворы проб и шкалы стандартов можно не сливать с силикагеля.
Расчет см. выше.
ЛИТЕРАТУРА
1. Алексеева М.В. Определение метилметакрилата. В кн.: Определение атмосферных загрязнений. М., «Медгиз», 1963.
2. Алексеева М.В. Определение метилового спирта. В кн.: Определение атмосферных загрязнений. М., «Медгиз», 1963.
3. Деяпова Е.В. Определение тетрагидрофурана. В кн.: Методы определения вредных веществ в воздухе. М., «Медицина», 1966.
4. Ефремова Г.П. Определение фенола. В сб.: Предельно допустимые концентрации атмосферных загрязнений. М., «Медицина», 1955, в. 2.
5. Кузьмичева М.Н. Определение сложных эфиров в воздухе. Гиг. труда и профзаболеваний, 1961, 5.
6. Масленников А.С. Определение малых количеств циклогексанола в водных растворах и в воздухе. Заводская лаборатория, 1954, 1.
7. Пинигина И.А. Раздельное определение алифатических спиртов в воздухе с помощью хроматографии на бумаге. Гиг. и сан., 1965, 11.
8. Пинигина И.А. О колориметрическом определении паров спиртов в воздухе. Гиг. и сан., 1966, 1.
9. Селина И.А. Определение вторичного пропилового (изопропилового) спирта в воздухе. Гиг. и сан., 1962, 12.
10. Хрусталева В.А. Определение фенола в атмосферном воздухе. Гиг. и сан., 1962, 10.
11. Хрусталева В.А., Осокина С.К. Раздельное определение винилацетата и 2-этилгексилакрилата в присутствии дибутилмалеината в воздухе. Гиг. и сан., 1970, 4.
Глава XI
АЛЬДЕГИДЫ, КЕТОНЫ
ФОРМАЛЬДЕГИД
Бесцветный газ с резким запахом, хорошо растворимый в воде; 35 — 40% раствор формальдегида называется формалином.
Температура кипения формальдегида — 21 °C.
Легко полимеризуется, образуя параформальдегид.
Оказывает раздражающее действие на верхние дыхательные пути и глаза. При попадании растворов формалина на кожу могут возникать дерматиты. Формальдегид обладает и общей токсичностью.
Предельно допустимые концентрации: максимальная разовая 0,035 мг/м3; среднесуточная 0,035 мг/м3.
ОПРЕДЕЛЕНИЕ ФОРМАЛЬДЕГИДА С ХРОМОТРОПОВОЙ КИСЛОТОЙ [1, 6]
Принцип определения
При взаимодействии формальдегида с хромотроповой кислотой образуется соединение, окрашенное в фиолетовый цвет. Интенсивность окраски пропорциональна количеству формальдегида.
Чувствительность определения при фотометрическом определении — 0,2 мкг в анализируемом объеме раствора.
Определению мешают: сероводород с 0,03 мг/м3, двуокись азота и метиловый спирт с 0,3 мг/м3, сернистый газ с 3 мг/м3, аммиак с 0,5 мг/м3, ацетон с 6 мг/м3.
Реактивы
1. Силикагель КСК, КСМ, промытый соляной кислотой, водой до нейтральной реакции, высушенный и активированный при 200 °C в течение 30 мин. Регенерация силикагеля осуществляется тем же способом.
2. Стандартный раствор формальдегида. 5 мл водного раствора формальдегида (40% раствор формалина) доводят в мерной колбе емкостью 250 мл до метки водой и определяют содержание формальдегида в этом растворе. Для этого 5 мл раствора помещают в коническую колбу емкостью 200 мл с притертой пробкой, приливают 20 мл 0,1 н. раствора йода и по каплям вносят 30% раствор едкого натра до появления устойчивой бледно-желтой окраски. Колбу оставляют на 10 мин., затем осторожно подкисляют раствор 2,5 мл соляной кислоты (1:5), оставляют на 10 мин. в темноте и оттитровывают избыток йода 0,1 н. раствором тиосульфата натрия. Когда раствор станет светло-желтым, прибавляют несколько капель 0,5% раствора крахмала. Предварительно устанавливают количество тиосульфата, расходуемое на титрование 20 мл 0,1 н. раствора. По разности между количеством тиосульфата, израсходованным на контрольное титрование, и избытком йода, не вошедшим в реакцию с формальдегидом, устанавливают количество йода, которое пошло на окисление формальдегида, 1 мл 0,1 н. раствора йода соответствует 1,5 мг формальдегида.
Установив содержание формальдегида в 1 мл раствора, соответствующим разведением водой готовят исходный стандартный раствор формальдегида с содержанием 100 мкг/мл. Раствор проверяют титрометрически.
Рабочий стандартный раствор с содержанием 1 мкг/мл формальдегида готовят в день анализа разбавлением исходного стандартного раствора в 100 раз дистиллированной водой.
3. Соляная кислота, приготовленная в отношении 1:5.
4. Йод, 0,1 н. раствор.
5. Тиосульфат, 0,1 н. раствор.
6. Серная кислота, 9 М раствор. К 530 мл воды осторожно приливают 470 мл серной кислоты удельного веса 1,80 — 1,82.
7. Хромотроповая кислота (1,8-диоксинафталин-3,6-дисульфокислота), 0,5 г кислоты растворяют в 10 мл воды. Хранят раствор в холодильнике, он устойчив в течение 3 — 5 дней, при пожелтении для использования не пригоден.
В колбу емкостью 50 мл вносят 2 мл раствора и раствор доводят до метки 15 М серной кислотой.
8. Серная кислота 15 М. К 100 мл воды осторожно при охлаждении приливают 900 мл серной кислоты удельного веса 1,80 — 1,82.
Отбор пробы
а) Для определения разовой концентрации воздух со скоростью 1 — 2 л/мин. протягивают через неподвижный слой силикагеля, помещенный в трубку (см. рис. 6). Продолжительность отбора 20 — 30 мин.
При концентрациях формальдегида в воздухе, превышающих допустимые, отбор проб можно проводить в 6 мл воды, помещенных в поглотительные приборы с пористой пластинкой: N 1, со скоростью до 0,5 л/мин.
б) Для определения среднесуточной концентрации воздух протягивают через слой силикагеля со скоростью до 1 л/мин. в течение суток непрерывно. Отбор проб допустимо проводить 6 — 12 раз с перерывами в 2 — 4 часа в течение 30 мин. в один и тот же поглотительный прибор.
Ход анализа
Силикагель переносят в колориметрическую пробирку, добавляют 5 мл дистиллированной воды и нагревают при 50 °C в течение 30 мин. (при закрытой пробке). Периодически перемешивают, добавляют к пробе 2 мл раствора хромотроповой кислоты, пробирки помещают в кипящую водяную баню на 30 мин.
Одновременно готовят стандартную шкалу, при этом наносят стандартный раствор на силикагель (табл. 73).
Таблица 73
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
|
Рабочий стандартный раствор, мл |
0 |
0,5 |
0,7 |
1,0 |
2 |
3 |
5 |
|
Дистиллированная вода, мл |
4,5 |
4,3 |
4 |
3 |
2 |
0 |
|
|
Содержание формальдегида, мкг |
0 |
0,5 |
0,7 |
1,0 |
2 |
3 |
5 |
Во все пробирки шкалы добавляют раствор хромотроповой кислоты и повторяют аналогичные операции.
По охлаждении через 30 — 40 мин. сравнивают интенсивность окраски проб с окраской шкалы.
При отборе пробы в жидкую поглотительную среду берут на анализ по 2 мл раствора из каждого поглотительного прибора.
Добавляют по 2 мл хромотроповой кислоты и помещают в кипящую водяную баню на 30 — 40 мин. По охлаждении растворы доливают до 5 мл 9 М раствором серной кислоты, помещают в кюветы с толщиной слоя 1 см и фотометрируют при длине волны 570 нм по сравнению с контролем, который готовят аналогично и одновременно с пробой. Содержание формальдегида определяют по заранее построенному графику. Для построения калибровочного графика готовят шкалу стандартов (табл. 74).
Таблица 74
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
|
Рабочий стандартный раствор, мл |
0 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
2,0 |
|
Поглотительный раствор, мл |
2 |
1,8 |
1,6 |
1,4 |
1,2 |
1 |
0 |
|
Содержание формальдегида, мкг |
0 |
0,2 |
0,4 |
0,6 |
0,8 |
1 |
2 |
Все пробирки шкалы обрабатывают аналогично пробе, измеряют оптическую плотность и строят график.
Расчет см. выше.
Примечания. 1. Очистка хромотроповой кислоты. Хромотроповую кислоту растворяют без нагревания в дистиллированной воде, фильтруют и выпаривают на водяной бане до начала кристаллизации. Охлаждают, отсасывают, промывают небольшим количеством холодной воды, а затем спиртом. Очищенную хромотроповую кислоту сушат в вакуум-эксикаторе и хранят в темной склянке.
2. Раздельное определение формальдегида и метилового спирта см. выше.
ОПРЕДЕЛЕНИЕ ФОРМАЛЬДЕГИДА
С СОЛЯНОКИСЛЫМ ФЕНИЛГИДРАЗИНОМ [3]
Принцип определения
При взаимодействии формальдегида с солянокислым фенилгидразином в присутствии окислителя в щелочной среде образуется соединение, окрашенное в красный цвет. Интенсивность окраски пропорциональна количеству формальдегида.
Чувствительность определения 0,2 мкг в анализируемом объеме раствора.
Аммиак и фенол не мешают определению, если присутствуют в растворе в пятикратном количестве по отношению к формальдегиду.
Реактивы
1. Поглотительный раствор, 50% изопропиловый спирт.
2. Ферроцианид калия, 5% раствор.
3. Солянокислый фенилгидразин, 5% раствор. Навеску соли растворяют в горячей воде и раствор фильтруют.
4. Едкий натр, 10% раствор.
5. Остальные реактивы см. выше.
Отбор пробы
Воздух со скоростью 0,5 — 1 л/мин. протягивают через поглотительный прибор с пористой пластинкой N 1, содержащий 6 мл поглотительного раствора.
Ход анализа
В колориметрическую пробирку вносят 5 мл пробы, приливают 0,2 мл 5% раствора солянокислого фенилгидразина, перемешивают и оставляют на 15 мин. Затем вносят по 0,1 мл 5% раствора ферроцианида калия и взбалтывают. Через 10 мин. добавляют 1 мл 10% раствора едкого натра, взбалтывают и через 30 мин. фотометрируют в кювете с толщиной слоя 1 см при длине волны 490 нм по сравнению с контролем, который готовят одновременно и аналогично пробам. Содержание формальдегида в анализируемом растворе определяют по предварительно построенному калибровочному графику. Для построения калибровочного графика готовят шкалу стандартов (табл. 75).
Таблица 75
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
9 |
10 |
|
Рабочий стандартный раствор, мл |
0 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
— |
— |
— |
— |
— |
|
Исходный стандартный раствор, мл |
0 |
— |
— |
— |
— |
— |
0,02 |
0,04 |
0,06 |
0,08 |
0,1 |
|
Поглотительный раствор, мл |
5 |
4,8 |
4,6 |
4,4 |
4,2 |
4,0 |
4,98 |
4,96 |
4,94 |
4,92 |
4,9 |
|
Содержание формальдегида, мкг |
0 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
2,0 |
4,0 |
6,0 |
8,0 |
10,0 |
Все пробирки шкалы обрабатывают аналогично пробам. Измеряют оптическую плотность и строят график. Шкалой стандартов можно пользоваться и для визуального определения, ее готовят в колориметрических пробирках одновременно с пробами.
Расчет см. выше.
Примечание. При раздельном определении формальдегида и метилового спирта строят два калибровочных графика: один по формальдегиду и второй — по метиловому спирту после его окисления.
Пробу делят пополам. В первой половине определяют формальдегид по величине оптической плотности (Д) по графику 1, построенному по формальдегиду. Во второй половине пробы проводят окисление метилового спирта до формальдегида и также измеряют величину оптической плотности (Д1). Разность оптических плотностей (Д1 — Д) соответствует величине оптической плотности (Д2), т.е. интенсивности окраски, полученной за счет окислившегося метилового спирта. В соответствии со значением Д2 по калибровочному графику 2, построенному по метиловому спирту, находят концентрацию метилового спирта. Полученные результаты удваивают.
ФУРФУРОЛ
Бесцветная жидкость, быстро буреющая на воздухе, с запахом миндаля, температура кипения 161,7 °C, плотность при 20 °C 1,59. Фурфурол растворим в воде, спирте, эфире и других растворителях.
Фурфурол является нервным ядом, вызывает судороги и паралич.
Предельно допустимые концентрации: максимальная разовая и среднесуточная — 0,05 мг/м3.
ОПРЕДЕЛЕНИЕ ФУРФУРОЛА С АНИЛИНОМ [2]
Принцип определения
При взаимодействии фурфурола с анилином в уксуснокислой среде образуется дианилид оксиглутаконового альдегида, окрашивающий раствор в розовый цвет.
Чувствительность определения — 0,5 мкг в анализируемом объеме раствора.
Альдегиды, спирты, ацетон, хлориды, минеральные кислоты не мешают определению.
Реактивы
1. Поглотительный раствор — этанол и вода в отношении 1:1.
2. Стандартный раствор фурфурола. Готовят исходный раствор в мерной колбе емкостью 50 мл, в которую наливают 10 — 15 мл поглотительного раствора. Колбу взвешивают и в нее вносят 2 — 3 капли фурфурола и снова взвешивают. По разности двух взвешиваний устанавливают количество фурфурола. Объем жидкости в колбе доводят до метки поглотительным раствором, содержимое колбы перемешивают, вычисляют количество фурфурола в 1 мл раствора. Рабочий стандартный раствор с содержанием 10 мкг/мл получают в мерной колбе емкостью 50 мл, в нее вносят такое количество исходного раствора, в котором содержится 500 мкг фурфурола. Раствор доводят до метки поглотительным раствором.
3. Этанол 96°.
4. Уксусная кислота 80 — 100%, раствор.
5. Раствор анилина в уксусной кислоте:
в мерную колбу емкостью 50 мл помещают 5 мл анилина и до метки добавляют уксусную кислоту, раствор тщательно перемешивают.
Отбор пробы
Для определения разовой концентрации протягивают воздух со скоростью 0,5 л/мин. через поглотительный прибор с пористым фильтром N 1, содержащим 5 мл поглотительного раствора. Продолжительность отбора 20 — 30 мин. при охлаждении.
Ход анализа
В пробирку, в которую вливают 0,5 мл раствора анилина в уксусной кислоте, вносят 4 мл пробы. Содержимое пробирки тщательно перемешивают и через 10 мин. измеряют оптическую плотность в кювете толщиной слоя 1 см по сравнению с контролем, который готовят одновременно и аналогично пробе. Определение производят при длине волны 530 нм.
Содержание фурфурола в анализируемом объеме определяют по предварительно построенному калибровочному графику. Для построения графика готовят шкалу стандартов (табл. 76).
Таблица 76
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
|
Рабочий стандартный раствор, мл |
0 |
0,05 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
|
Поглотительный раствор, мл |
4 |
3,95 |
3,9 |
3,8 |
3,6 |
3,4 |
3,2 |
3,0 |
|
Содержание фурфурола, мкг |
0 |
0,5 |
1 |
2 |
4 |
6 |
8 |
10 |
Все пробирки шкалы обрабатывают аналогично пробе.
Шкалой стандартов можно пользоваться для визуального определения. Шкалу готовят в колориметрических пробирках одновременно с пробой.
Расчет см. выше.
АКРОЛЕИН
|
CH2 =CH·CHO |
Мол. вес 56,06 |
Бесцветная летучая жидкость с неприятным запахом, температура кипения 52,5 °C, температура плавления — 87,7 °C; плотность 0,84. Акролеин растворим в воде, хорошо растворяется в спирте и других органических растворителях. Образуется при неполном сгорании жиров, масел, глицерина, встречается в выхлопных газах, в литейных цехах и т.п. Вызывает резкое раздражение верхних дыхательных путей, глаз.
Предельно допустимые концентрации: максимальная разовая — 0,03 мг/м3.
ОПРЕДЕЛЕНИЕ АКРОЛЕИНА С 4-ГЕКСИЛРЕЗОРЦИНОМ [4, 8]
Принцип определения
При взаимодействии акролеина с 4-гексилрезорцином в присутствии хлорной или уксуснокислой ртути образуется соединение, окрашенное в голубой цвет.
Чувствительность определения 1 мкг акролеина в анализируемом объеме раствора.
Определению не мешают двуокись азота в количествах до 5 мкг, кротоновый альдегид до 2 мкг. Не мешают определению сернистый газ, ароматические углеводороды, фенолы, эфиры, кетоны.
Реактивы
1. Поглотительный раствор. Готовят смешением растворов 4-гексилрезорцина, хлорида или ацетата ртути, трихлоруксусной кислоты и этилового спирта в отношениях 1:2:50:50.
Поглотительный раствор устойчив в течение дня.
2. Исходный стандартный раствор готовят в этаноле с содержанием 100 мкг/мл. Рабочий стандартный раствор с содержанием 10 мкг/мл готовят соответствующим разведением исходного.
3. 4-Гексилрезорцин, 50% раствор в этаноле.
4. Хлорная ртуть, 3% раствор в этаноле.
5. Ацетат ртути (двузамещенная соль), 3% раствор в этаноле.
6. Трихлоруксусная кислота, насыщенный раствор. 100 г кислоты (х. ч.) растворяют в 10 мл дистиллированной воды при нагревании на водяной бане, затем объем доводят до 70 мл водой. Раствор употребляют свежеприготовленный.
7. Этиловый спирт, ректификат.
Отбор пробы
Для определения максимальной разовой концентрации воздух со скоростью 0,5 л/мин. протягивают через два поглотительных прибора с пористой пластинкой N 1, содержащих по 5 мл поглотительного раствора. Продолжительность отбора — 60 мин.
Ход анализа
Определение акролеина проводят в день отбора пробы или на следующий день. Пробу хранят в холодильнике. При анализе температура раствора должна быть 18 — 20 °C. Содержимое каждого поглотительного прибора доводят до первоначального объема этиловым спиртом, переносят в пробирки, нагревают в течение 30 мин. на водяной бане при 60 °C. По охлаждении через 15 мин. фотометрируют окрашенный раствор в кюветах с толщиной слоя 1 см при длине волны 605 нм по сравнению с контролем. Содержание акролеина в анализируемом объеме определяют по предварительно построенному калибровочному графику. Для построения калибровочного графика готовят шкалу стандартов (табл. 77).
Таблица 77
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
|
Рабочий стандартный раствор, мл |
0 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
|
Этиловый спирт, мл |
1,8 |
1,7 |
1,6 |
1,4 |
1,2 |
1,0 |
0,8 |
|
Содержание акролеина, мкг |
0 |
1 |
2 |
4 |
6 |
8 |
10 |
При приготовлении шкалы в каждую пробирку вносят 0,5 мл раствора 4-гексилрезорцина, 0,2 мл раствора хлорной ртути или 0,2 мл раствора ацетата ртути, 2,5 мл раствора трихлоруксусной кислоты.
После тщательного перемешивания пробирки помещают на 30 мин. в водяную баню, нагретую до 60 °C, фотометрируют и строят калибровочный график.
Шкалой стандартов можно пользоваться для визуального определения, ее готовят в колориметрических пробирках одновременно с пробами.
Расчет см. выше.
ОПРЕДЕЛЕНИЕ АКРОЛЕИНА ФЛУОРЕСЦЕНТНЫМ МЕТОДОМ [5]
Принцип определения
При взаимодействии акролеина с м-фенилендиамином образуется производное, которое выделяют из смеси в тонком слое силикагеля. Полученное соединение в ультрафиолете флуоресцирует голубым светом.
Чувствительность определения — 0,1 мкг акролеина в 5 мл элюата. Определению не мешают: предельные, непредельные, ароматические альдегиды. Кротоновый и уксусный альдегиды также дают флуоресцирующие соединения, но зоны локализаций этих соединений расположены на других уровнях на хроматограмме и определению акролеина не мешают.
Аппаратура
1. Флуорометр, лампа УФ-ПР К-4 или ПР К-2 со стеклом Вуда.
Реактивы
1. Поглотительный раствор. Гидроксиламин солянокислый, 0,25% раствор.
2. Исходный стандартный раствор акролеина готовят с содержанием 1 — 2 мг/мл в воде. Рабочий стандартный раствор готовят разбавлением исходного 0,25% раствором гидроксиламина. Рабочий стандартный раствор содержит 2 мкг/мл акролеина.
Исходный и рабочий растворы готовят перед употреблением.
3. М-Фенилендиамин, 0,5% раствор, приготовленный в 1 н. растворе соляной кислоты. Хранят в темном месте. Срок хранения 3 — 4 дня.
4. Соляная кислота, 5 н., 1 н. растворы.
5. Аммиак, 25% раствор.
6. Хлороформ.
7. Изопропиловый спирт.
8. Уксусная кислота, ледяная.
9. Гидроксиламин солянокислый, 0,25% раствор.
10. Спирт этиловый, ректификат, осушенный.
11. Системы растворителей: диметилформамид — бензол 1:2 или четыреххлористый углерод — этиловый спирт 1:5 или хлороформ — этиловый спирт 1:4 или этиловый спирт — хлороформ 4:1 по объему.
12. Силикагель КСМ, КСК, очищенный. Силикагель заливают соляной кислотой 1:1 и выдерживают в течение 18 — 20 ч, промывают водой. Добавляют азотной кислоты 1:1 и кипятят в течение 2 — 3 ч. Далее промывают проточной водопроводной водой, затем дистиллированной до нейтральной реакции промывных вод. Высушивают при 130 °C в течение 4 — 6 ч.
13. Кальций сернокислый (гипс), х.ч. или специально приготовленный. Для этого водный раствор хлористого кальция смешивают с эквимолярным количеством серной кислоты при 70 — 80 °C. Сульфат кальция отделяют, промывают водой до нейтральной реакции и высушивают при 120 °C.
14. Хроматографические пластинки «Silufol» или специально приготовленные. Для этого берут стеклянную пластинку размером 9 x 12 см, промывают хромовой смесью, дистиллированной водой и высушивают в вертикальном положении. Перед нанесением сорбционной массы пластину протирают этиловым спиртом. Берут 5 г силикагеля с зернами величиной 0,1 — 0,07 мм, смешивают с 0,5 г гипса и добавляют 10 мл воды. К полученной смеси добавляют еще 8 мл воды и тщательно перемешивают. 10 г сорбционной массы наносят на пластину и равномерно распределяют по поверхности. Высушивают в течение часа при 100 °C. Хранят над хлористым кальцием. Хроматографические пластины, обработанные люминесцирующим реактивом, употреблять не рекомендуется.
Отбор пробы
а) Для определения максимальной разовой концентрации акролеина воздух со скоростью 0,5 л/мин. протягивают через два последовательно соединенных поглотительных прибора Зайцева, наполненных по 5 мл 0,25% раствора солянокислого гидроксиламина. Продолжительность отбора 20 — 30 мин.
б) Для определения среднесуточной концентрации воздух протягивают через два последовательно соединенных поглотительных прибора Зайцева, наполненных по 10 мл 0,25% раствора солянокислого гидроксиламина, со скоростью 0,2 — 0,3 л/мин. 6 — 12 раз в одну и ту же поглотительную систему с перерывами в 2 — 4 ч.
Ход анализа
Жидкость из каждого поглотительного прибора переносят в пробирки. Добавляют по 1 мл 0,5% раствора м-фенилендиамина и по 1 мл 5 н. раствора соляной кислоты. Содержимое пробирок перемешивают и помещают в кипящую водяную баню на 15 мин. По охлаждении содержимое пробирок переносят в делительные воронки, вносят по 0,5 мл 25% раствора аммиака и по 5 мл хлороформа.
Смесь перемешивают в течение 1 — 2 мин. и нижний слой хлороформа фильтруют через бумажные фильтры в сухие пробирки или выпарительные чашки. Хлороформ удаляют на водяной бане при температуре не выше 60 °C. Сухие остатки обрабатывают 0,1 мл хлороформа и по 0,05 мл с помощью микропипетки наносят на стартовую линию на хроматографическую пластину. Предварительно на пластине намечают точки на расстоянии 1 см от края и на расстоянии 2,5 см друг от друга. После высыхания (удаления хлороформа) пластину помещают в хроматографическую камеру, в которую заранее вносят одну из систем растворителей. Смесь диметилформамида с бензолом или хлороформа с этиловым спиртом помещают в делительную воронку, перемешивают и вносят в хроматографическую камеру с тем расчетом, чтобы пластина погружалась в систему не более чем на 0,5 см. После того как линия фронта будет на расстоянии 1 см от верхнего края пластины, хроматограмму вынимают из камеры, высушивают на воздухе и просматривают в ультрафиолетовом свете (с фильтром N 3). Флуоресцирующие голубым светом зоны локализации, соответствующие акролеину, определяют по свидетелю, нанесенному на пластину одновременно с пробой (так как в разных системах растворителей и на разных пластинах Rf производных акролеина различна).
Участки силикагеля с флуоресцирующими производными акролеина снимают и помещают в пробирки. В пробирки вносят по 6 мл 1 н. раствора соляной кислоты, перемешивают в течение 1 — 2 мин. и центрифугируют. Интенсивность флуоресценции элюатов измеряют на флуориметре с желтым светофильтром. Содержание акролеина находят по калибровочному графику. Для построения калибровочного графика готовят шкалу стандартов в соответствии с табл. 78.
Таблица 78
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
|
Рабочий стандартный раствор, мл |
0 |
0,05 |
0,1 |
0,2 |
0,4 |
0,5 |
0,6 |
0,7 |
|
Поглотительный раствор, мл |
5 |
4,95 |
4,9 |
4,8 |
4,6 |
4,5 |
4,4 |
4,3 |
|
Содержание акролеина, мкг |
0 |
0,1 |
0,2 |
0,4 |
0,8 |
1,0 |
1,2 |
1,4 |
Полученные таким образом растворы 1 — 2…7 обрабатывают аналогично пробе. Для построения калибровочного графика сухие остатки растворяют в 0,1 мл хлороформа и в количестве по 0,05 мл наносят на пластины, что соответствует 0,05 — 0,1 — 0,2 — 0,4 — 0,5 — 0,6 и 0,7 мкг акролеина. Раствор 1 н. соляной кислоты устанавливают на 6% по шкале флуориметра; этот раствор служит контролем. Затем измеряют интенсивность флуоресценции всех элюатов, показание контроля вычитают. График строят на основании средних данных из 5 — 6 шкал.
Расчет анализа см. выше.
АЦЕТОН
Бесцветная жидкость с характерным запахом. Температура кипения 56,24 °C, плотность при 20 °C 0,791. Хорошо растворим в воде, спирте, эфире и других органических растворителях. Ацетон является хорошим растворителем жира, резины, воска, нитроцеллюлозы. Смесь паров ацетона с воздухом обладает взрывчатыми свойствами.
Ацетон действует как наркотик, поражает центральную нервную систему.
Предельно допустимая концентрация: максимальная разовая и среднесуточная 0,35 мг/м3.
ОПРЕДЕЛЕНИЕ АЦЕТОНА С САЛИЦИЛОВЫМ АЛЬДЕГИДОМ [11]
Принцип определения
При взаимодействии ацетона с салициловым альдегидом в щелочной среде образуется соединение, окрашивающее раствор в желтый цвет, по интенсивности которого определяют содержание ацетона.
Чувствительность определения — 1 мкг в анализируемом объеме раствора. Спирты, альдегиды до концентрации 7 мг/л не мешают определению.
Реактивы
1. Силикагель КСК, величина зерен 0,5 — 2 мм. Силикагель обрабатывают соляной кислотой, разбавленной водой в отношении 1:1 при нагревании в течение 3 — 4 ч, затем промывают водой, высушивают при 105 °C и активируют при 200 °C в течение 30 мин. Силикагель проверяют на отсутствие ацетона. Бывший в употреблении силикагель регенерируют, промывают и активируют.
2. Поглотительный раствор — дистиллированная вода.
3. Стандартный раствор ацетона: готовят исходный раствор в мерной колбе емкостью 50 мл, в которую влито 15 — 20 мл воды. Колбу взвешивают, вносят в нее 2 — 3 капли свежеперегнанного ацетона и снова взвешивают. По разности двух взвешиваний устанавливают количество ацетона в колбе. Раствор встряхивают, доводя до метки водой. Высчитывают количество ацетона в 1 мл.
Рабочий стандартный раствор с содержанием 10 мкг/мл готовят в мерной колбе емкостью 100 мл, в нее вносят такое количество исходного раствора, которое содержит 1000 мкг/мл ацетона. Раствор доводят до метки водой.
Применяют только свежеприготовленный раствор.
4. Салициловый альдегид 2,18 мл свежеперегнанного альдегида (температура кипения 193 °C), вносят в мерную колбу емкостью 50 мл, в которую влито 10 — 20 мл этанола, и добавляют до метки этанолом.
5. Этанол 96°.
6. Едкое кали, 50% раствор.
Отбор пробы
а) Для определения разовой концентрации протягивают воздух через два поглотительных прибора Института имени Ф.Ф. Эрисмана, к которым присоединяют прибор для улавливания переброшенных частиц. Каждый прибор содержит по 3 мл силикагеля, скорость отбора через «кипящий слой» силикагеля — 5 л/мин.
При отборе пробы в жидкую поглотительную среду воздух протягивают со скоростью 0,3 л/мин., через поглотительный прибор с пористым фильтром N 1, содержащий 4 мл воды. Продолжительность отбора 20 — 30 мин.
б) Для определения среднесуточной концентрации воздух протягивают в кипящий слой силикагеля со скоростью 1 — 2 л/мин. непрерывно в течение суток. Допустимо отбор пробы проводить 6 — 12 раз с перерывами в 2 — 4 ч по 30 мин. в один и тот же поглотительный прибор.
Ход анализа
Силикагель пересыпают в пробирки с притертыми пробками из каждого прибора отдельно. Переброшенные зерна из уловителя пересыпают в пробирки второго прибора.
Силикагель в каждой пробирке заливают 6 мл воды и помещают на водяную баню, нагретую до 40 °C, на 30 мин., производя встряхивание раствора через 5 — 10 мин.
По охлаждении раствора 2 мл пробы переводят в пробирку и наливают 1 мл 50% раствора едкого кали и 0,5 мл салицилового альдегида. Содержимое пробирки встряхивают и помещают на 25 мин. в водяную баню, нагретую до 70°.
После охлаждения раствора проводят определение ацетона, сравнивая интенсивность окраски пробы со шкалой стандартов (табл. 79).
Таблица 79
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
|
Рабочий стандартный раствор, мл |
0 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
|
Вода, мл |
2 |
1,9 |
1,8 |
1,6 |
1,4 |
1,2 |
1,0 |
|
Содержание ацетона, мкг |
0 |
1 |
2 |
4 |
6 |
8 |
10 |
Все пробирки шкалы обрабатывают аналогично пробе одновременно с ней.
При отборе проб в воду отбирают 2 мл пробы и обрабатывают аналогично, используя ту же шкалу стандартов.
Расчет см. выше.
ЦИКЛОГЕКСАНОН
Бесцветная маслянистая жидкость с запахом мяты, температура кипения 155,4 °C, плотность пара по отношению к воздуху 3,4. Почти не растворима в воде, хорошо растворима в ряде органических растворителей.
Циклогексанон раздражающе действует на слизистые оболочки глаз, горла и носа.
Предельно допустимые концентрации: максимальная разовая и среднесуточная 0,04 мг/м3.
ОПРЕДЕЛЕНИЕ ЦИКЛОГЕКСАНОНА С ФУРФУРОЛОМ [7]
Принцип определения
При взаимодействии циклогексанона и фурфурола в щелочной среде образуется фурфурилиденциклогексанон. Последний, вступая в соединение с серной кислотой, окрашивает раствор в малиновый цвет, по интенсивности которого определяют содержание циклогексанона.
Чувствительность определения — 0,5 мкг в анализируемом объеме раствора.
Другие кетоны мешают определению.
Реактивы
1. Поглотительный раствор — этанол, разбавленный водой в отношении 1:1.
2. Стандартный раствор циклогексанона: готовят исходный раствор в мерной колбе емкостью 50 мл, в которую вливают 5 — 10 мл поглотительного раствора, и колбу взвешивают. Затем в колбу вносят 0,1 — 0,2 мл циклогексанона и вновь взвешивают. Содержимое колбы доводят до метки поглотительным раствором и взбалтывают. По разности двух взвешиваний устанавливают количество циклогексанона в колбе и высчитывают его содержание в 1 мл.
Рабочий стандартный раствор с содержанием 10 мкг/мл циклогексанона готовят в мерной колбе емкостью 50 мл. В нее вносят такое количество исходного раствора, в котором содержится 500 мкг циклогексанона. Объем жидкости в колбе доводят до метки поглотительным раствором.
3. Этанол, 96°.
4. Фурфурол свежеперегнанный, из него готовят 0,3% водный раствор; он устойчив в течение 2 — 3 недель.
5. Натр едкий, 25% раствор.
6. Серная кислота, уд. вес 1,84.
Отбор пробы
Для определения разовой концентрации воздух протягивают со скоростью 0,5 л/мин. через поглотительный прибор с пористым фильтром N 1, содержащий 3 мл поглотительного раствора. Продолжительность отбора 20 — 30 мин. При отборе поглотительный прибор помещают в баню со льдом.
Ход анализа
В пробирку помещают 2 мл пробы, добавляют 0,2 мл 0,3% раствора фурфурола и 0,2 мл 25% раствора едкого натра. Раствор в пробирке встряхивают и оставляют в покое в течение часа. По истечении этого времени в пробирку добавляют 3 мл серной кислоты уд. веса 1,84. Содержимое пробирки встряхивают и по охлаждении измеряют оптическую плотность в кювете с толщиной слоя 1 см по сравнению с контролем, который готовят одновременно и аналогично пробе. Определение проводят при длине волны 555 нм. Содержание циклогексанона в анализируемом объеме определяют по предварительно построенному калибровочному графику. Для построения графика готовят шкалу стандартов.
Все пробирки шкалы обрабатывают аналогично пробе.
Шкалой стандартов можно пользоваться для визуального определения, ее готовят в колориметрических пробирках одновременно с пробой (табл. 80).
Расчет см. выше.
Таблица 80
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
|
Рабочий стандартный раствор, мл |
0 |
0,05 |
0,07 |
0,09 |
0,1 |
0,15 |
0,2 |
0,3 |
|
Поглотительный раствор, мл |
2 |
1,95 |
1,93 |
1,91 |
1,9 |
1,85 |
1,8 |
1,7 |
|
Содержание циклогексанона, мкг |
0 |
0,5 |
0,7 |
0,9 |
1,0 |
1,5 |
2,0 |
3,0 |
АЦЕТОФЕНОН
(Метилфенилкетон, ацетилбензол)
Жидкость без цвета, с резко выраженным запахом. Температура плавления 19,7 °C; температура кипения 202,3 °C. Не растворим в воде, хорошо растворим в этаноле, эфире и других органических растворителях. Ацетофенон токсичен.
Предельно допустимые концентрации: максимальная разовая и среднесуточная — 0,003 мг/м3.
ОПРЕДЕЛЕНИЕ АЦЕТОФЕНОНА С М-ДИНИТРОБЕНЗОЛОМ [10]
Принцип определения
При взаимодействии ацетофенона с М-динитробензолом в щелочной среде образуется продукт, окрашенный в розовый цвет.
Чувствительность определения — 2 мкг в анализируемом объеме раствора.
Ацетон мешает определению в количествах, превышающих 100 мкг. Бензол, изопропилбензол, гидроперекись изопропилбензола, ![]() -метилстирол, стирол, фенол не мешают определению.
-метилстирол, стирол, фенол не мешают определению.
Реактивы
1. Силикагель КСК, МСК, КСМ, МСМ с зернами величиной 0,25 — 2 мм, обработанных, как указано выше. Каждую партию силикагеля проверяют на отсутствие ацетофенона.
2. Стандартный раствор ацетофенона. Исходный стандартный раствор готовят путем растворения точной навески ацетофенона в этаноле. Рабочий стандартный раствор с содержанием 10 мкг ацетофенона в 1 мл готовят соответствующим разбавлением исходного, перед употреблением.
3. М-динитробензол, 1% раствор в этаноле готовят в малом объеме (10 мл), раствор устойчив в течение суток.
А. Калий едкий, 30% раствор.
Отбор пробы
А) Воздух со скоростью до 10 л/мин. протягивают в «кипящий» слой силикагеля. Для этого по 3 мл силикагеля помещают в два последовательно соединенных поглотительных прибора Яворовской или НИИ гигиены имени Ф.Ф. Эрисмана, соединенных с поглотителем для задержания переброшенных частиц. Отбор проб проводят при охлаждении. Отбирают не менее часа.
Б) Для определения среднесуточной концентрации воздух со скоростью 5 — 10 л/мин. протягивают в кипящий слой силикагеля непрерывно в течение суток.
Допустимо проводить отбор 6 — 12 раз с перерывами в 4 или 2 ч.
При отборе поглотительный прибор помещают в охладительную смесь.
Ход анализа
Содержимое поглотительных приборов переносят в отдельные пробирки с притертыми пробками. Во вторую пробирку помещают переброшенные частицы силикагеля.
Заливают силикагель по 4 мл этанола. Периодически встряхивают в течение 30 мин. Добавляют по 0,2 мл раствора М-динитробензола, по 0,3 мл раствора едкого кали и по 0,5 мл воды.
Содержимое пробирок перемешивают. Одновременно готовят шкалу стандартов. Для этого в серию пробирок вносят по 3 мл силикагеля, который употреблен при анализе, наносят на силикагель рабочий стандартный раствор, перемешивают. Одновременно готовят контроль (табл. 81).
Таблица 81
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
9 |
|
Рабочий стандартный раствор, мл |
0 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
1,2 |
1,5 |
2,0 |
2,5 |
|
Этанол, мл |
По 4 мл |
|||||||||
|
Содержание ацетофенона, мкг |
0 |
2 |
4 |
6 |
8 |
10 |
12 |
15 |
20 |
25 |
В каждую пробирку шкалы и в контроль вносят по 0,2 мл раствора М-динитробензола и другие реактивы аналогично пробе.
Через час сравнивают интенсивность окраски в пробах с окраской стандартной шкалы в растворах над силикагелем.
Расчет см. выше.
ДИКЕТЕН
|
|
Мол. вес 84,0 |
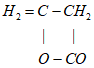
Бесцветная жидкость с острым запахом, растворимая в органических растворителях, не растворимая в воде. Температура кипения 127,4 °C, плотность 1,089. Дикетен весьма реакционноспособен, легко подвергается полимеризации.
Пары дикетена вызывают резкое раздражение глаз и дыхательных путей. Характерными являются бронхиты с пневматическими очагами, отеки легких. Предельно допустимые концентрации: максимальная разовая — 0,007 мг/м3.
ОПРЕДЕЛЕНИЕ ДИКЕТЕНА С РЕЗОРЦИНОМ [9]
Принцип определения
Продукт реакции дикетена с серной кислотой, взаимодействуя с резорцином, дает сине-фиолетовую люминесценцию при облучении ультрафиолетовым излучением.
Чувствительность определения — 0,02 мкг в анализируемом объеме раствора.
Определению мешает кетен.
Ацетон до 170 мкг, уксусный ангидрид до 70 мкг, уксусная кислота, метан, этилен, окись и двуокись углерода не мешают определению.
Реактивы
1. Поглотительный раствор: серная кислота, приготовленная в отношении 8:2. Для приготовления 100 мл раствора в колбу вводят 20 мл дистиллированной воды, а затем добавляют 80 мл концентрированной серной кислоты. Полученный раствор охлаждают и проверяют на отсутствие люминесценции.
2. Дикетен перегнанный.
Перегонку дикетена производят под вакуумом при остаточном давлении 50 мм рт. ст. Стабилизируют дикетен сернокислым цинком из расчета 1 г на 100 мл. Хранить дикетен следует в холодильнике.
3. Стандартные растворы дикетена. Для приготовления исходного раствора дикетена в мерную колбу емкостью 25 — 50 мл наливают 5 — 10 мл дистиллированной воды и взвешивают на аналитических весах.
В колбу вносят 1 — 2 капли дикетена и снова взвешивают. Объем в колбе доводят до метки водой и хорошо перемешивают. По разности в весе определяют содержание дикетена в 1 мл раствора. Из исходного раствора готовят стандартный раствор с содержанием 2 мкг/мл, а из него рабочий стандартный раствор с содержанием 0,2 мкг/мл. Рабочий стандартный раствор готовят на серной кислоте, приготовленной в отношении 8:2.
Стандартный раствор устойчив в течение 5 суток при хранении в холодильнике.
3. Серная кислота, уд. вес 1,84.
4. Резорцин. Если резорцин загрязнен или окрашен, его очищают возгонкой. Для этого 2 — 3 г резорцина помещают на часовое стекло, которое покрывают другим таким же стеклом. Между стеклами прокладывают фильтровальную бумагу с большим количеством отверстий. Возгонку резорцина проводят на песчаной бане при 115 — 120 °C. При возгонке верхнее стекло охлаждают фильтровальной бумагой, смоченной водой. Образующиеся между фильтровальной бумагой и верхним стеклом прозрачные кристаллы резорцина собирают и хранят в склянке из темного стекла с притертой пробкой.
5. 1% водный раствор резорцина. Раствор хранят в темной склянке с притертой пробкой, он устойчив в течение 1 — 2 мес.
6. Сульфат цинка, прокаленный для удаления влаги.
7. Дистиллированная вода, нелюминесцирующая под ультрафиолетовым светом.
Отбор пробы
Для определения разовой концентрации воздух со скоростью 0,1 л/мин. протягивают через поглотительный прибор Полежаева, содержащий 1 мл поглотительного раствора, в течение 20 — 30 мин.
Ход анализа
Готовят шкалу стандартов в пробирках из нелюминесцирующего стекла (табл. 82).
Таблица 82
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
9 |
10 |
|
Рабочий стендартный раствор, мл |
0 |
0,1 |
0,2 |
0,3 |
0,4 |
0,5 |
0,6 |
0,7 |
0,8 |
0,9 |
1,0 |
|
Поглотительный раствор, мл |
1 |
0,9 |
0,8 |
0,7 |
0,6 |
0,5 |
0,4 |
0,3 |
0,2 |
0,1 |
0 |
|
Содержание дикетена, мкг |
0 |
0,02 |
0,04 |
0,06 |
0,08 |
0,1 |
0,12 |
0,14 |
0,16 |
0,18 |
0,20 |
Одновременно в поглотители с пробами и в пробирки шкалы добавляют по 0,1 мл 1% раствора резорцина и хорошо перемешивают. Содержимое поглотителей переливают в пробирки из нелюминесцирующего стекла и через 15 мин. сопоставляют интенсивность свечения пробы со шкалой стандартов на установке для люминесцентного визуального анализа (лампа ПРК-4, светофильтр УФС-3). Шкалой стандартов можно пользоваться в течение 4 — 5 ч. Контроль не должен давать люминесценции. При наличии в нем люминесценции следует проверить чистоту пробирок и реактивов.
Расчет см. выше.
ЛИТЕРАТУРА
1. Алексеева М.В. Определение формальдегида. В кн.: Определение атмосферных загрязнений. М., «Медгиз», 1963.
2. Алексеева М.В. Определение фурфурола. В кн.: Определение атмосферных загрязнений. М., «Медгиз», 1963.
3. Бензина Г.И. Фотометрический метод определения формальдегида в воздухе. Гиг. и сан., 1968, 6.
4. Ипатова С.А. К вопросу определения акролеина с 4-гексилрезорцином. Гиг. и сан., 1971, 1.
5. Ипатова С.А., Деянова Е.В. Определение акролеина в воздухе флюоресцентным методом. Гиг. и саи., 1973, 10.
6. Качмар Е.Г., Хрусталева В.А. Применение зерненных сорбентов для отбора проб атмосферного воздуха. Гиг. и сан., 1969, 9.
7. Масленников А.С. Определение циклогексанона в воздухе. Заводская лаборатория, 1954, 1.
8. Новый спектрофотометрический метод определения акролеина в дымовых газах и атмосфере. Реферат (пер. с англ. Л.И. Вагранской). Гиг. и сан., 1963, 9.
9. Определение дикетена в воздухе. Технические условия на методы определения вредных веществ в воздухе. В. 5. М., «Медицина», 1968.
10. Хрусталева В.А. Определение ацетофенона в воздухе. Гиг. и сан., 1961, 12.
11. Шихваргер Ф.Д. Количественное определение ацетона в атмосферном воздухе по реакции с салициловым альдегидом. Заводская лаборатория, 1951, т. 17, с. 11.
Глава XII
АНГИДРИДЫ, ОРГАНИЧЕСКИЕ КИСЛОТЫ
МАЛЕИНОВЫЙ АНГИДРИД
|
|
Мол. вес 98,06 |
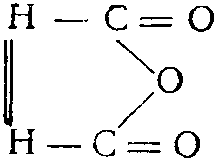
Бесцветное кристаллическое вещество с температурой плавления 52 — 53 °C, температурой кипения 202 °C, обладает резким запахом, растворим в воде, спирте, ацетоне. Применяется при получении полиэфирных смол, при диеновых синтезах. Обладает местным раздражающим действием, вызывает конъюнктивит, раздражение верхних дыхательных путей, кожи.
Предельно допустимые концентрации: максимальная разовая — 0,2 мг/м3, среднесуточная — 0,05 мг/м3.
ОПРЕДЕЛЕНИЕ ПАРОВ МАЛЕИНОВОГО АНГИДРИДА ОКИСЛЕНИЕМ [3]
Принцип определения
Метод основан на окислении малеиновой кислоты, образующейся в водной среде из малеинового ангидрида, до винной кислоты. Окисление проходит по месту двойной связи, при этом фиолетовый цвет раскисляющегося перманганата переходит в желтовато-бурый.
Чувствительность определения — 0,5 мкг в анализируемом объеме раствора. Определению мешают непредельные соединения: фталиевый ангидрид, эпихлоргидрин; гликоли не мешают определению.
Реактивы
1. Поглотительный раствор — дистиллированная вода.
2. Стандартный раствор малеинового ангидрида с содержанием 100 мкг/мл. Из него разбавлением в 10 раз готовят рабочий стандартный раствор с содержанием 10 мкг/мл.
3. Перманганат калия, 0,04% раствор.
Отбор пробы
Для определения разовой концентрации воздух со скоростью 0,2 — 0,3 л/мин. протягивают через два поглотительных прибора с пористой пластинкой N 1, содержащих по 3 мл дистиллированной воды.
Продолжительность отбора 20 — 30 мин.
Ход анализа
Из каждого поглотительного прибора 2 мл пробы берут на анализ. Одновременно готовят шкалу стандартов (табл. 83).
Таблица 83
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
|
Рабочий стандартный раствор, мл |
0 |
0,05 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
|
Вода дистиллированная, мл |
2 |
1,95 |
1,90 |
1,8 |
1,6 |
1,4 |
1,2 |
1,0 |
|
Содержание малеинового ангидрида, мкг |
0 |
0,5 |
1,0 |
2,0 |
4,0 |
6,0 |
8,0 |
10,0 |
Во все пробирки шкалы и пробы добавляют по 0,1 мл 0,04% раствора марганцевокислого калия. Через 5 — 10 мин. проводят сравнение окраски пробы со шкалой.
Расчет см. выше.
Примечание. При наличии в воздухе аэрозоля малеинового ангидрида отбор проб проводят на фильтр АФА.
ОДНООСНОВНЫЕ КАРБОНОВЫЕ КИСЛОТЫ C1 — C9
CнH2нO2
Прозрачные бесцветные жидкости. Кислоты C1 — C3 обладают резким запахом, C — C — неприятным прогорклым запахом. Кислоты C4 — C9 хорошо растворимы в воде, но с увеличением молекулярного веса растворимость в воде уменьшается. Все кислоты хорошо растворимы в этаноле и эфире.
Токсические свойства кислот по силе и характеру воздействия неодинаковы. Муравьиная и уксусная кислоты сильно раздражают верхние дыхательные пути; эти кислоты, попадая на кожу, вызывают ожоги, пропионовая кислота вызывает умеренное раздражение кожи. Раздражающее действие среднемолекулярных кислот C1 — C9 выражено слабо.
Попадая в организм, кислоты вызывают изменение внутренних органов, крови, сосудисто-сердечной и нервной системы и др. Некоторые свойства карбоновых кислот представлены в табл. 84.
Таблица 84
НЕКОТОРЫЕ ФИЗИЧЕСКИЕ СВОЙСТВА И ПРЕДЕЛЬНО ДОПУСТИМЫЕ
КОНЦЕНТРАЦИИ КАРБОНОВЫХ КИСЛОТ
|
Название |
Формула |
Молекулярный вес |
Удельный вес (20°) |
Температура кипения |
ПДК мг/м3 |
|
|
максимальная разовая |
среднесуточная |
|||||
|
Муравьиная |
C1H2O2 |
46,03 |
1,2200 |
100, 7 |
||
|
Уксусная |
C2H4O2 |
60,05 |
1,0490 |
118,1 |
0,2 |
0,06 |
|
Пропионовая |
C3H6O2 |
74,08 |
0,9934 |
141,1 |
||
|
Масляная |
C4H8O2 |
88, 10 |
0,9587 |
163,5 |
0,015 |
0,01 |
|
Валериановая |
C5H10O2 |
102,14 |
0,9420 |
186,3 |
0,03 |
0,01 |
|
Капроновая |
C6H12O2 |
116,16 |
0,9290 |
205,0 |
0,01 |
0,005 |
|
Энантовая |
C7H14O2 |
130,19 |
0,9184 |
2283,0 |
||
|
Каприловая |
C8H16O2 |
144,22 |
0,9100 |
237,5 |
||
|
Пеларгоновая |
C9H18O2 |
158,24 |
0,9055 |
254,0 |
ОПРЕДЕЛЕНИЕ ОБЩЕГО КОЛИЧЕСТВА ОДНООСНОВНЫХ
КАРБОНОВЫХ КИСЛОТ C1 — C9 [2]
Принцип определения
При взаимодействии кислот с метанолом в присутствии серной кислоты образуются эфиры, которые, вступая в реакцию с гидроксиламином в щелочной среде, превращаются в гидроксаматы. Последние в кислой среде, реагируя с хлорным железом, переходят в железогидроксамовый комплекс, окрашивая раствор в буро-розовый цвет.
Чувствительность фотометрического определения — 10 — 20 мкг (C1 — C3) и (C4 — C9) соответственно в анализируемом объеме раствора.
Чувствительность визуального определения — 2 мкг в анализируемом объеме раствора.
Минеральные кислоты, ацетон, динил, спирты, сложные летучие эфиры жирных кислот определению не мешают.
Реактивы
1. Силикагель СМ для отбора проб, величина зерен от 0,5 до 2 мм. Силикагель предварительно обрабатывают соляной кислотой, разбавленной водой в отношении 1:1 при нагревании до 60 — 80°, периодически помешивая силикагель. Пожелтевшую кислоту несколько раз заменяют свежим раствором. Обработку заканчивают, когда раствор соляной кислоты остается бесцветным. Затем силикагель отмывают от кислоты водой, сушат при 105 °C и активируют при 200 — 250 °C в течение 20 мин. Обработанный силикагель регенерируют описанным выше методом.
2. Стандартный раствор кислот. Готовят исходный стандартный раствор из каждой кислоты в мерных колбах емкостью 25 мл. В колбы вливают 10 — 5 мл этанола, колбы взвешивают, вносят в каждую по 5 — 10 капель кислоты и снова взвешивают, объем раствора доводят до метки этанолом. По разности двух взвешиваний устанавливают в каждой колбе содержание кислоты и высчитывают количество кислоты в 1 мл раствора. Рабочий стандартный раствор должен содержать все исследуемые кислоты по 100 мкг/мл каждой кислоты. Для этого в мерную колбу емкостью 50 мл вносят такое количество исходного раствора каждой кислоты, в котором содержится 5 мг. Объем колбы доводят до метки этанолом.
3. Серная кислота, уд. вес 1,84%, 50% в метаноле.
4. Гидроксиламин солянокислый, 4 н. раствор.
5. Едкий натр, 4 и 0,05 н. раствор свежеприготовленный.
6. Соляная кислота, 0,5 и 4 н. раствор.
7. Хлорное железо окисное 1,6% раствор в 0,5 н. растворе соляной кислоты.
8. Этанол, обработанный щелочью и перегнанный.
9. Метанол осушенный. Обезвоживание метанола проводят металлическим магнием.
Отбор пробы
а) Для определения максимальной разовой концентрации воздух протягивают со скоростью 5 — 6 л/мин. через два последовательно соединенных поглотительных прибора — Яворовской и НИИ гигиены имени Ф.Ф. Эрисмана, наполненных 2 г силикагеля, в «кипящий» слой. Продолжительность отбора 40 мин.
б) Для определения среднесуточной концентрации воздух со скоростью 2 — 3 л/мин. протягивают через силикагель в течение суток непрерывно. Допустимо отбор проводить 6 — 12 раз с перерывами в 4 — 2 ч по 20 — 30 мин.
Ход анализа
Силикагель переносят в пробирки из каждого поглотительного прибора отдельно. Переброшенные зерна силикагеля присоединяют к силикагелю из контрольного поглотительного прибора.
Пробу, помещенную в пробирку, заливают 5 мл этанола, встряхивают и оставляют на 60 мин., периодически встряхивая. По истечении этого времени для анализа берут 3 мл и помещают в фарфоровый тигель. Затем кислоту переводят в соли путем титрования пробы 0,05 н. раствором едкого натра в присутствии одной капли фенолфталеина.
Тигель помещают в кипящую водяную баню для удаления этанола. Вместе с этанолом испаряются эфиры, если они присутствуют в пробе.
Сухой остаток подсушивают в эксикаторе или в сушильном шкафу при 80° (эта операция обязательна).
Осадок в тигле постепенно растворяют в 1,5 мл метанола в три приема по 0,5 мл каждый раз, переводя пробу в колориметрическую пробирку. В пробу вносят одну каплю серной кислоты уд. веса 1,84. Содержимое пробирки встряхивают и оставляют на 30 мин., периодически встряхивая жидкость.
Через 30 мин. в пробирку вливают 0,2 мл 4 н. раствора солянокислого гидроксиламина и 0,6 мл 4 н. раствора едкого натра. Раствор встряхивают и оставляют на 15 мин. По истечении указанного времени раствор нейтрализуют 4 н. соляной кислотой по фенолфталеину. Затем вносят 2,5 мл 1,6% раствора хлорида железа окисного. Раствор встряхивают, через 20 — 30 мин. измеряют оптическую плотность раствора в кювете с толщиной слоя 1 см при длине волны 490 нм по сравнению с контролем, который готовят одновременно и аналогично пробе. Содержание кислоты в анализируемом объеме раствора определяют по калибровочному графику, который строят по шкале стандартов (табл. 85).
Таблица 85
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
|
Рабочий стандартный раствор, мл |
0 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
1,2 |
1,5 |
|
Метанол, мл |
1,5 |
1,4 |
1,3 |
1,1 |
0,9 |
0,7 |
0,5 |
0,3 |
0 |
|
Содержание кислот, мкг |
0 |
10 |
20 |
40 |
60 |
80 |
100 |
120 |
150 |
Все пробирки шкалы обрабатывают аналогично пробе.
При визуальном определении проводят те же операции, как описано выше, но сухой остаток после подсушивания в сушильном шкафу растворяют в 0,75 мл метанола.
В пробу вносят одну каплю 50% раствора серной кислоты в метаноле и оставляют пробу на 30 мин. для полного образования эфира. Временами раствор встряхивают. Через 30 мин. в тигель вливают 0,1 мл 4 н. раствора гидроксиламина и 0,4 мл 4 н. раствора едкого натра. Раствор перемешивают и через 15 мин. переливают в колориметрическую пробирку. В тигель вливают 1,5 мл 1,6% раствора хлорного железа и его ополаскивают этим раствором. Затем раствор переливают из тигля в пробирку с пробой. Проба окрашивается в буро-розовый цвет.
Через 10 — 20 мин. интенсивность окраски пробы сравнивают со шкалой стандартов, которую готовят одновременно с пробой (табл. 86).
Таблица 86
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
|
Рабочий стандартный раствор, мл |
0 |
0,02 |
0,05 |
0,10 |
0,20 |
0,30 |
0,40 |
0,50 |
|
Метанол, мл |
0,75 |
0,73 |
0,70 |
0,65 |
0,55 |
0,45 |
0,35 |
0,25 |
|
Содержание кислот, мкг |
0 |
2 |
5 |
10 |
20 |
30 |
40 |
50 |
Все пробирки шкалы обрабатывают аналогично пробе.
Расчет см. выше.
РАЗДЕЛЬНОЕ ОПРЕДЕЛЕНИЕ КАРБОНОВЫХ КИСЛОТ
(C1 — C9) ПРИ ПОМОЩИ БУМАЖНОЙ ХРОМАТОГРАФИИ [1]
Принцип определения
Метод основан на переведении кислот в натриевые соли, затем в соответствующие гидроксамовые кислоты, которые разделяют на бумаге нисходящим методом в двух системах растворителей — бутанол, уксусная кислота, вода в соотношении 4:1:5 (для C1 — C4) и бензол, муравьиная кислота, вода в соотношении 1:1:1 (для C5 — C9).
После проявления хроматограммы производные кислот обнаруживаются в виде фиолетовых пятен.
Количественное определение проводят визуально путем сравнения интенсивности окраски пятен проб с окраской «свидетелей», а также фотометрически, после окисления элюированных гидроксамовых кислот до азотистой кислоты и определения последней с сулфаниловой кислотой и ![]() -нафтиламином. Минимальные количества, открываемые на хроматограммах, соответствуют 3 мкг для каждой из кислот C1 — C4 и 5 мкг для каждой из кислот C5 — C9. Определению не мешают минеральные кислоты, спирты, сложные эфиры жирных кислот групп C1 — C9, формальдегид, кетоны, амины C7 — C9, динил.
-нафтиламином. Минимальные количества, открываемые на хроматограммах, соответствуют 3 мкг для каждой из кислот C1 — C4 и 5 мкг для каждой из кислот C5 — C9. Определению не мешают минеральные кислоты, спирты, сложные эфиры жирных кислот групп C1 — C9, формальдегид, кетоны, амины C7 — C9, динил.
Аппаратура
1. Хроматографическая камера, представляющая собой стеклянный цилиндр высотой 50 — 55 см, диаметром 20 — 30 см с хорошо пришлифованной крышкой (см. рис. 26).
2. Стеклянная лодочка.
3. Теплоэлектровентилятор.
4. Стеклянный распылитель.
5. Пробирки высотой 60 мм, диаметр 8 мм с делениями на 0,3 и 0,5 мл.
6. Микропипетки емкостью 0,1 и 0,2 мл с делениями 0,01 мл.
Реактивы
1. Стандартные растворы жирных кислот (муравьиной, уксусной, пропионовой, масляной, валерьяновой, капроновой, энантовой, каприловой, пеларгоновой) с содержанием 1 мг/мл метанола.
2. Силикагель марки СМ с зернами размером 0,5 — 2 мм.
Предварительно силикагель обрабатывают 50% (по объему) раствором соляной кислоты при 60 — 80 °C. Порции кислоты меняют до тех пор, пока она не будет бесцветной. Затем силикагель промывают водой и 2 — 3 раза — 50% (по объему) раствором азотной кислоты, отмывают горячей водой от ионов хлора и азотной кислоты, сушат при 105 °C и активируют в течение 30 мин. при 200 — 300 °C.
3. Метанол (метиловый спирт), обезвоженный металлическим магнием. Для этого в круглодонную колбу с обратным холодильником помещают 0,5 г йода, 5 г магния и 50 — 75 мл метанола. Смесь нагревают на водяной бане с помощью закрытой плитки в вытяжном шкафу. Горелки, расположенные поблизости, должны быть выключены, так как в результате реакции выделяется водород. (Если при этом не происходит бурного выделения водорода, добавляют еще 0,5 г йода.) Смесь нагревают до тех пор, пока весь магний не превратится в метилат магния. Затем прибавляют еще 900 мл метанола и смесь кипятят с обратным холодильником в течение 30 мин. Метанол отгоняют, предохраняя от влаги. Хранят в склянке с хорошо притертой пробкой.
4. Этанол 96°, обработанный едким кали; для этого к 100 мл спирта добавляют 20 г едкого кали, перемешивают и оставляют стоять 2 — 3 ч, после чего спирт отгоняют при 78°.
5. Серный эфир, для наркоза.
6. Едкий натр 0,05 н. водно-спиртовой (1:1) раствор.
7. Фенолфталеин 0,5% спиртовой раствор.
8. Едкий натр, 5% раствор в этиловом спирте.
9. Щелочной раствор гидроксиламина. К 10 мл 5% раствора гидроксиламина прибавляют 20 мл 5% раствора едкого натра и ставят на 5 — 10 мин. в сосуд со льдом. При этом выпадает обильный осадок хлористого натрия, который удаляют фильтрованием.
Данный раствор готовят в день употребления.
10. Соляная кислота, 2 н. раствор в этиловом спирте.
11. Хлорное железо, 0,5% раствор в этиловом спирте с добавлением 0,2 мл соляной кислоты (уд. вес 1,19).
12. Уксусная кислота, ледяная.
13. Муравьиная кислота.
14. Бутанол.
15. Бензол.
16. Щавелевая кислота, 1% раствор.
17. Серная кислота, уд. вес 1,84.
18. Натрий уксуснокислый, 0,5% раствор.
19. Йод, 0,1 М раствор в ледяной уксусной кислоте.
20. Сульфаниловая кислота, 0,5 г растворяют в 150 мл 30% уксусной кислоты.
21. ![]() -Нафтиламин, 0,1 г растворяют в 20 мл воды при нагревании до образования лиловых пятен на дне колбы. Раствор над осадком сливают в темную склянку и в нее вливают 110 мл 10% раствора уксусной кислоты.
-Нафтиламин, 0,1 г растворяют в 20 мл воды при нагревании до образования лиловых пятен на дне колбы. Раствор над осадком сливают в темную склянку и в нее вливают 110 мл 10% раствора уксусной кислоты.
22. Тиосульфат натрия, 0,1 н. раствор.
23. Хроматографическая бумага «Медленная», «Быстрая».
Отбор пробы
Для определения максимальной разовой концентрации воздух протягивают через два последовательно соединенных поглотительных прибора — Яворовской или НИИ гигиены имени Ф.Ф. Эрисмана, наполненные по 2 мл силикагеля. Протягивают 50 — 150 л исследуемого воздуха со скоростью 5 л/мин.
Ход анализа
1. Получение гидроксамовых кислот
а) Обработка проб.
После отбора пробы силикагель переносят в пробирку с притертой пробкой и заливают 5 мл этанола. Кислоты извлекают при комнатной температуре в течение 60 мин. при периодическом и интенсивном встряхивании.
Для анализа берут 3 мл прозрачного раствора, переносят в фарфоровый тигель и титруют 0,05 н. раствором едкого натра до слабо-розового окрашивания по фенолфталеину. Растворитель удаляют при 90 — 95 °C. Остаток натриевых солей жирных кислот подсушивают в эксикаторе или сушильном шкафу (80 °C). Сухой остаток солей смывают 1 мл метанола (дробными порциями), переносят в колориметрическую пробирку на 10 мл, прибавляют по 1 капле серной кислоты и оставляют на 30 мин. при комнатной температуре для образования эфиров. Через 30 мин. в каждую пробирку добавляют по 2 мл щелочного раствора гидроксиламина, заранее рассчитанное количество 5% раствора едкого натра для нейтрализации 1 капли серной кислоты и 5 мл эфира.
Смесь оставляют на 40 мин. при комнатной температуре для образования гидроксаматов. Через 40 мин. раствор нейтрализуют 2 н. раствором соляной кислоты по фенолфталеину до обесцвечивания малиновой окраски, содержимое взбалтывают и затем дают осесть осадку. Раствор над осадком сливают через складчатый фильтр, стараясь, чтобы на фильтр попало как можно меньше осадка. Затем снова к осадку в пробирке добавляют 1 — 1,5 мл эфира и повторяют извлечение гидроксамовых кислот. И так до 3 раз. Фильтрат переносят в выпарительную чашку и удаляют растворитель в вытяжном шкафу при комнатной температуре. Остаток смывают 0,3 мл метанола (дробными порциями) и переносят в круглодонную приборку с делением 0,3 мл, доводят этанолом до метки; 0,05 — 0,1 мл этого раствора используют для анализа кислот группы C1 — C4 и 0,05 — 0,1 мл для анализа кислот группы C5 — C9.
б) Приготовление «свидетелей».
Стандартные растворы кислот группы C1 — C4 смешивают по 0,2 мл каждой. Так же поступают со стандартными растворами кислот C5 — C9. Дальнейшая обработка стандартных растворов кислот проводится аналогично обработке проб воздуха.
После удаления растворителя полученные гидроксамовые кислоты растворяют в 1 мл метанола, что соответствует содержанию 200 мкг/мл каждой из кислот.
2. Разделение производных кислот
Для разделения производных кислот C1 — C4 используется хроматографическая бумага марок «Медленная» или «Быстрая» Ленинградской фабрики. На листе промытой бумаги проводят линию старта в 6,5 см от края, на которой намечают четыре точки с расстоянием 3,5 см друг от друга.
С помощью микропипетки в одну точку наносят 0,1 мл раствора смеси соответствующих «свидетелей» кислот группы C1 — C4, в три другие — пробы в количестве по 0,1 мл; при этом диаметр пятен на линии старта не должен превышать 5 — 6 мм.
С целью ускорения нанесения проб на бумагу следует пользоваться теплоэлектровентилятором.
Кислоты группы C5 — C9 делят на бумаге марки «Медленная».
После нанесения проб бумагу помещают в лодочку с подвижным растворителем, укрепленную в верхней части хроматографической камеры (предварительно насыщенную парами неподвижного и подвижного растворителей). Разделение идет нисходящим методом. Для разделения кислот C1 — C4 берут систему растворителей: бутанол, уксусную кислоту, воду в соотношении 4:1:5 помещают в делительную воронку, в которой при интенсивном встряхивании бутанол насыщается водой и уксусной кислотой. После разделения слоев нижний водный слой используют для насыщения камеры, для чего внутренние стенки ее покрывают фильтровальной бумагой, которая пропитывается растворителем. Концы бумаги не должны касаться друг друга, оставляя окно. Насыщать камеру следует не менее чем за 6 ч до начала анализа.
Верхний бутанольный слой из делительной воронки переносят в лодочку и используют в качестве подвижной фазы. Разделение кислот C1 — C4 идет в течение 18 — 20 ч. В качестве подвижной фазы при разделении кислот группы C5 — C9 используют бензол, насыщенный водой и муравьиной кислотой в соотношении 1:1:1. Разделение идет 5 — 6 ч. Разделение считается законченным, когда подвижный растворитель продвинется по бумаге на 25 — 30 см от линии старта. Далее хроматограмму извлекают из камеры, сушат в вытяжном шкафу при комнатной температуре в течение 30 — 60 мин. и проявляют.
3. Обнаружение производных кислот
Хроматограмму проявляют, опрыскивая из пульверизатора 0,5% раствором хлорного железа. Производные кислот проявляют в виде фиолетовых пятен на слабо-желтом фоне, которые расположены на хроматограмме последовательно в порядке увеличения числа углеродных атомов. Путем сравнения расположения на хроматограмме пятен проб с пятнами «свидетелей» делают выводы о наличии в пробах тех или иных кислот, так как производные одной и той же кислоты должны находиться на одинаковом расстоянии от линии старта (рис. 27).
Рис. 27. Хроматограмма карбоновых кислот гр. C1 — C4
(муравьиной, уксусной, пронионовой, масляной)
4. Количественное определение кислот группы
C1 — C9 методом визуальной оценки
Готовят растворы «свидетелей» с различным содержанием производных кислот. Для этого используют «свидетели», приготовленные согласно описанному выше способу. После получения гидроксамовых производных кислот растворитель испаряют, а сухие остатки производных растворяют в 1 мл этанола. Из полученных исходных растворов готовят серии разведений, как показано в табл. 87, 88.
Таблица 87
ПРИГОТОВЛЕНИЕ СЕРИИ «СВИДЕТЕЛЕЙ»
ДЛЯ ОПРЕДЕЛЕНИЯ КИСЛОТ C1 — C4
|
Исходный раствор гидроксамовых кислот, мл |
Этанол, мл |
Соответствующее количество каждой кислоты, мкг |
|
|
в 1 мл |
в 0,1 мл |
||
|
1,0 |
0 |
200,0 |
20,0 |
|
0,15 |
0,05 |
150,0 |
15,0 |
|
0,1 |
0,1 |
100,0 |
10,0 |
|
0,05 |
0,15 |
50,0 |
5,0 |
|
0,03 |
0,17 |
30,0 |
3,0 |
Таблица 88
ПРИГОТОВЛЕНИЕ СЕРИИ «СВИДЕТЕЛЕЙ» ДЛЯ ОПРЕДЕЛЕНИЯ
КИСЛОТ C5 — C9
|
Исходный раствор гидроксамовых кислот, мл |
Этанол, мл |
Соответствующее количество каждой кислоты, мкг |
|
|
в 1 мл |
в 0,1 мл |
||
|
0,5 |
0 |
400,0 |
40,0 |
|
0,1 |
0,1 |
200,0 |
20,0 |
|
0,1 |
0,3 |
100,0 |
10,0 |
|
0,05 |
0,35 |
50 |
5,0 |
Для определения кислот на лист хроматографической бумаги в разные точки наносят по 0,1 мл раствора «свидетелей». Затем «свидетели» подвергаются хроматографическому анализу в тех же условиях, что и пробы. На проявленных хроматограммах обнаруживаются пятна, различные по величине и интенсивности окраски и соответствующие 3 — 5 — 10 — 15 — 20 мкг каждой из кислот C1 — C4 и 5 — 10 — 20 — 40 мкг кислот C5 — C9.
Количественное определение кислот (после разделения их производных и проявления хроматограмм) методом визуальной оценки основано на том, что размер пятен и интенсивность окраски производных пропорциональны содержанию кислот. Сравнивая интенсивность окраски пятен проб с окраской «свидетелей», определяют содержание каждой кислоты в пробе.
5. Фотометрическое определение кислот C1 — C4
При фотометрическом определении содержания кислот в пробе предварительно готовят серию стандартов, как описано выше, т.е. подвергают хроматографическому разделению «свидетелей», соответствующих 3 — 5 — 10 — 15 — 20 мкг каждой из кислот C1 — C4. После проявления хроматограмм вырезают каждое пятно из бумаги, измельчают, помещают в пробирку и заливают 6 мл 0,5% раствора уксуснокислого натрия. Содержимое пробирки оставляют на 30 мин. при комнатной температуре, время от времени слегка встряхивают. Через 30 мин. 5 мл прозрачного раствора переносят в другую пробирку, добавляют 2 капли 0,1 М раствора йода и 0,3 мл раствора сульфаниловой кислоты.
Смесь помещают на 10 мин. в темное место. Через 10 мин. избыток йода связывают 0,1 н. раствором тиосульфата и добавляют 0,3 мл раствора ![]() -нафтиламина. Через 5 мин. величину оптической плотности раствора, окрашенного в малиновый цвет, измеряют при длине волны 520 нм.
-нафтиламина. Через 5 мин. величину оптической плотности раствора, окрашенного в малиновый цвет, измеряют при длине волны 520 нм.
Опыты повторяют 4 — 5 раз и по средним полученным данным строят градуировочный график зависимости оптической плотности элюатов от концентрации вещества.
Такие графики строят для каждой кислоты: муравьиной, масляной, уксусной, пропионовой.
Для определения содержания кислот в пробах после их хроматографического анализа последние обрабатывают так же, как и «свидетели». Величину оптической плотности окрашенного раствора проб измеряют на фотоколориметре и с помощью графиков находят количество кислоты в пробе.
Расчет см. на выше.
ЛИТЕРАТУРА
1. Абрамова Ю.В. Раздельное определение одноосновных карбоновых кислот в воздухе от C1 до C9 с помощью хроматографии на бумаге. Гиг. и сан., 1968, 6.
2. Абрамова Ю.В. К вопросу колориметрического определения жирных кислот группы C1 — C9 в воздухе. Гиг. и сан., 1969, 11.
3. Пименова З.М. Определение малеинового ангидрида. В кн.: М.С. Быховская, С.Л. Гинзбург и др. Методы определения вредных веществ в воздухе. «Медицина». М., 1966.
Глава XIII
АМИНОСОЕДИНЕНИЯ
ДИЭТИЛАМИН
Бесцветная жидкость с резким запахом, смешивается с водой, растворима в органических растворителях. Температура кипения 55,5 °C, плотность 0,710 (18 °C). Оказывает действие на центральную нервную систему, а также обладает раздражающими свойствами.
Предельно допустимые концентрации диэтиламина: максимальная разовая 0,05 мг/м3, среднесуточная 0,05 мг/м3.
ОПРЕДЕЛЕНИЕ ДИЭТИЛАМИНА С СЕРОУГЛЕРОДОМ [4]
Принцип определения
При взаимодействии диэтиламина с сероуглеродом и солями меди образуется диэтилдитиокарбамат меди, окрашенный в желто-зеленый цвет. Интенсивность окраски пропорциональна количеству диэтиламина.
Чувствительность определения — 0,5 мкг диэтиламина в анализируемом объеме раствора. Моноэтиламин до 50 мкг, а также триэтиламины и аммиак определению не мешают.
Реактивы
1. Поглотительный раствор: этиловый спирт, 96% ректификат.
2. Стандартный раствор диэтиламина. Исходный стандартный раствор готовят следующим образом. В мерную колбу емкостью 25 — 50 мл вливают 10 — 15 мл этилового спирта, взвешивают, вносят несколько капель диэтиламина и снова взвешивают. Объем жидкости в колбе доводят до метки этиловым спиртом и хорошо перемешивают. По разности в весе определяют количество диэтиламина и вычисляют его содержание в 1 мл раствора. Рабочий стандартный раствор с содержанием 10 мкг/мл диэтиламина готовят соответствующим разведением исходного раствора спиртом.
3. Сероуглерод, 10% спиртовой раствор (по объему), 1 мл сероуглерода вносят в небольшое количество спирта в пробирку с притертой пробкой и доводят объем до 10 мл спиртом.
4. Ацетат меди, спиртовой раствор, 10 мг соли растворяют в 0,5 мл воды и добавляют 9,5 мл этилового спирта.
5. Серная кислота, уд. вес 1,84.
6. Аммиак, спиртовой раствор, 1 мл 25% раствора аммиака разбавляют 9 мл этилового спирта. К 4 мл воды в конической колбе добавляют 1 мл спиртового раствора аммиака и оттитровывают 0,1 н. раствором соляной кислоты в присутствии индикатора метилового красного. Контрольный опыт обязателен. 1 мл 0,1 н. раствора соляной кислоты соответствует 1,7 мг аммиака. Готовят раствор, содержащий 7 — 10 мг/мл.
Растворы сероуглерода, ацетата меди и аммиака устойчивы в течение 2 — 3 дней.
Отбор пробы
Для определения разовой концентрации воздух со скоростью 0,5 л/мин. протягивают через поглотительный прибор с пористой пластинкой N 1, содержащий 2 мл 96% этилового спирта. Продолжительность отбора 20 — 30 мин. Поглотительные приборы следует охлаждать, при испарении доводить до первоначального объема.
Ход анализа
1 мл пробы вносят в колориметрическую пробирку. Одновременно готовят шкалу стандартов (табл. 89).
Таблица 89
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
0 |
1 |
2 |
3 |
4 |
5 |
6 |
|
Рабочий стандартный раствор, мл |
0 |
0,05 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
|
Этиловый спирт, мл |
1 |
0,95 |
0,9 |
0,8 |
0,6 |
0,4 |
0,2 |
|
Содержание диэтиламина, мкг |
0 |
0,5 |
1 |
2 |
4 |
6 |
8 |
В каждую пробирку шкалы и пробы прибавляют 0,1 мл спиртового раствора аммиака, 0,1 мл 10% спиртового раствора сероуглерода и 0,1 мл спиртового раствора ацетата меди и оставляют на 6 — 7 мин. По истечении этого времени добавляют по 0,25 мл серной кислоты уд. веса 1,84. После добавления каждого реактива раствор перемешивают. Интенсивность окраски пробы сравнивают со шкалой стандартов.
Расчет см. выше.
ТРИЭТИЛАМИН
Бесцветная жидкость с аммиачным запахом. Плотность 0,728 (20°), температура кипения 89,5°.
Триэтиламин очень хорошо растворим в органических растворителях.
Предельно допустимые концентрации: максимальная разовая и среднесуточная 0,14 мг/м3.
ОПРЕДЕЛЕНИЕ ТРИЭТИЛАМИНА С БРОМФЕНОЛОВЫМ СИНИМ [4]
Принцип определения
При взаимодействии триэтиламина с бромфеноловым синим образуется соединение, окрашенное в желтый цвет.
Чувствительность определения — 1 мкг триэтиламина в анализируемом объеме раствора. Моноэтиламин, диэтиламин, аммиак определению не мешают. Другие третичные амины мешают определению.
Реактивы
1. Поглотительный раствор: 0,01 н. раствор соляной кислоты.
2. Стандартный раствор триэтиламина. Исходный раствор готовят в мерной колбе емкостью 25 мл, в которую вносят 2 — 3 мл 0,01 н. раствора соляной кислоты, взвешивают, вносят 2 — 3 капли триэтиламина и снова взвешивают. Объем жидкости доводят до метки 0,01 н. раствором соляной кислоты и рассчитывают содержание триэтиламина в 1 мл. Рабочий стандартный раствор с содержанием 10 мкг/мл готовят соответствующим разбавлением исходного раствора 0,01 н. раствором соляной кислоты.
3. Раствор бромфенолового синего. 0,1 г реактива растворяют в 10 мл этилового спирта и разбавляют водой до объема 100 мл.
4. Ацетатный буферный раствор с pH 3,5 — 4,5. 50 мл 1 н. раствора ацетата натрия смешивают с 40 мл 1 н. раствора соляной кислоты и доводят водой до объема 250 мл.
Отбор пробы
а) Для определения максимальной разовой концентрации воздух со скоростью 1 л/мин. протягивают через поглотительный прибор с пористой пластинкой N 1, содержащий 6 мл поглотительного раствора. Продолжительность отбора 20 — 30 мин.
б) Для определения среднесуточной концентрации воздух со скоростью 0,3 — 0,5 л/мин. протягивают через поглотительный прибор с пористым фильтром, содержащий 6 мл поглотительного раствора непрерывно в течение суток. Перемешивают, охлаждают в воде со льдом. Добавляют 4 мл хлороформа, вновь охлаждают.
Ход анализа
К 5 мл пробы добавляют 1 мл ацетатного буферного раствора и 5 мл бромфенолового синего. Перемешивают и охлаждают в течение 5 мин. в воде со льдом. Добавляют 4 мл хлороформа, вновь охлаждают 5 мин. Смесь помещают в делительную воронку, перемешивают в течение 15 мин. Слой хлороформа переносят в кювету с толщиной слоя 10 мм, фотометрируют при 420 нм по сравнению с контролем, который готовят аналогично пробе.
Содержание триэтиламина определяют по калибровочному графику.
Для построения графика готовят шкалу стандартов (табл. 90).
Таблица 90
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
|
Рабочий стандартный раствор, мл |
0 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
2,0 |
|
Поглотительный раствор, мл |
5 |
4,9 |
4,8 |
4,6 |
4,4 |
4,2 |
4,0 |
3,0 |
|
Содержание триэтиламина, мкг |
0 |
1 |
2 |
4 |
6 |
8 |
10 |
20 |
Все пробирки шкалы обрабатывают аналогично пробе. Шкалой можно пользоваться для визуального определения, ее готовят одновременно с пробами.
Расчет см. выше.
ГЕКСАМЕТИЛЕНДИАМИН (ГМД)
|
NH2(CH2)6NH2 |
Мол. вес 116,2 |
Твердое вещество, обладающее резким неприятным запахом, легко растворимое в воде и органических растворителях. Температура плавления 39 °C, температура кипения 196 °C. ГМД легко возгоняется, является сильным основанием. Оказывает действие на нервную систему и вызывает изменения в крови.
Предельно допустимые концентрации гексаметилендиамина: максимальная разовая 0,001 мг/м3, среднесуточная 0,001 мг/м3.
ОПРЕДЕЛЕНИЕ ГЕКСАМЕТИЛЕНДИАМИНА
С 2,4-ДИНИТРОХЛОРБЕНЗОЛОМ [7]
Принцип определения
При взаимодействии гексаметилендиамина с 2,4-динитрохлорбензолом образуется соединение, окрашенное в желтый цвет. Интенсивность окраски пропорциональна количеству гексаметилендиамина.
Чувствительность определения — 0,3 мкг в анализируемом объеме раствора.
Определению мешает аммиак с 5 мг в анализируемом объеме, а также первичные, вторичные и некоторые третичные амины.
Реактивы
1. Поглотительный раствор: серная кислота, 0,1 н. раствор.
2. Стандартные растворы ГМД.
Исходный раствор с содержанием 100 мкг/мл готовят растворением 0,05 г ГМД (бесцветного) в 500 мл 0,1 н. раствора серной кислоты. Рабочий стандартный раствор, содержащий 1 мкг/мл ГМД, готовят разведением исходного раствора в 100 раз 0,1 н. раствором серной кислоты.
3. Карбонат натрия (Na2CO3·10H2O), 36% раствор. Готовят при нагревании на водяной бане.
4. 2,4-Динитрохлорбензол, 5% спиртовой раствор. Готовят при нагревании на водяной бане.
5. Соляная кислота, уд. вес 1,19.
6. Хлороформ.
Отбор пробы
Воздух со скоростью 2 л/мин. протягивают через два поглотительных прибора с пористой пластинкой N 1 (диаметр фильтра 20 — 25 мм), содержащих по 10 мл 0,1 н. раствора серной кислоты.
Ход анализа
Для анализа из каждого поглотительного прибора берут 9 мл пробы и вносят в конические колбы с притертыми пробками емкостью 25 — 30 мл; в таких же колбах готовят шкалу стандартов (табл. 91).
Таблица 91
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
|
Рабочий стандартный раствор, мл |
0 |
0,3 |
0,6 |
1,2 |
2,4 |
3,6 |
|
Поглотительный раствор, мл |
9 |
8,7 |
8,4 |
7,8 |
6,6 |
5,4 |
|
Содержание ГМД, мкг |
0 |
0,3 |
0,6 |
1,2 |
2,4 |
3,6 |
Во все колбы шкалы и проб вливают по 0,6 мл 36% раствора карбоната натрия и по 1,2 мл 5% раствора динитрохлорбензола. Колбы без пробок помещают в кипящую водяную баню на 30 мин., закрепив их в штативе. Колбы охлаждают и вливают по 0,3 мл соляной кислоты уд. веса 1,19 и по 0,5 мл (точно) хлороформа. Колбы закрывают пробками и ставят на 20 мин. в аппарат для встряхивания и полного экстрагирования. После экстрагирования растворы переливают в конические центрифужные пробирки и колориметрируют. Шкала в закрытых пробирках устройства в течение 2 нед.
Расчет см. выше.
АРОМАТИЧЕСКИЕ АМИНЫ
АНИЛИН
Бесцветная маслянистая жидкость с характерным запахом, быстро темнеющая на свету, плотность при 20 °C 1,022, температура кипения 184,4 °C. Анилин хорошо растворим в эфире, спирте, ацетоне, бензоле, хлороформе и других органических растворителях. При 20 °C 3,4 г анилина растворяются в 100 мл воды. Анилин обладает основными свойствами, с кислотами образует соли.
Анилин действует на центральную нервную систему и является ядом крови.
Предельно допустимые концентрации: максимальная разовая 0,05 мг/м3, среднесуточная 0,03 мг/м3.
ОПРЕДЕЛЕНИЕ АНИЛИНА ПО ИНДОФЕНОЛУ [1]
Принцип определения
При взаимодействии анилина и фенола с хлорамином в щелочной среде образуется индофенол, который окрашивает раствор в синий цвет.
Чувствительность определения — 0,5 мкг в анализируемом объеме раствора.
Ароматические амины и аммиак мешают определению.
Реактивы
1. Поглотительный раствор: 0,01 н. раствор серной кислоты готовят разбавлением 0,1 н. серной кислоты в 10 раз.
2. Силикагель АСМ, КСМ, КСК с зернами величиной 0,25 — 2 мм, промытый водой при кипячении, высушенный при 105° и активированный при 200° в течение 30 мин., проверенный на отсутствие анилина.
3. Стандартный раствор анилина. Исходный стандартный раствор готовят растворением точной навески анилина в 0,01 н. растворе серной кислоты. Рассчитывают содержание анилина в 1 мл раствора.
Рабочий стандартный раствор с содержанием 10 мкг/мл готовят из исходного соответствующим разбавлением его 0,01 н. раствором серной кислоты.
4. Хлорамин, 4% раствор, растворяют в воде, нагретой до 40 — 50° и фильтруют.
5. Фенол, 3% раствор. Готовят из бесцветного свежеперегнанного фенола.
6. Натр едкий, 2% и 0,1 н. растворы.
7. Серная кислота, 0,1 н. раствор.
Отбор пробы
а) Для определения разовой концентрации протягивают воздух со скоростью 1 л/мин. через поглотительный прибор с пористым фильтром N 1, содержащий 4 мл поглотительного раствора. Отбор проб можно проводить в «кипящий» слой силикагеля. Для этого 3 мл силикагеля помещают в два последовательно присоединенных поглотительных прибора — Яворовской и Института имени Ф.Ф. Эрисмана, соединенных с поглотителем для улавливания переброшенных частиц. Отбирают пробу со скоростью до 10 л/мин. Продолжительность отбора 20 — 30 мин.
б) Для определения среднесуточной концентрации непрерывно в течение суток воздух со скоростью 0,3 л/мин. протягивают через поглотительный прибор, содержащий 4 мл поглотительного раствора. При наполнении прибора отмечают уровень жидкости, который периодически добавляют водой до первоначального. Допустимо проводить отбор 6 — 12 раз с перерывами в 2 — 4 ч в один и тот же поглотительный прибор.
Для отбора пробы также используют и силикагель, скорость отбора до 5 л/мин.
Ход анализа
Силикагель переносят отдельно из каждого поглотительного прибора в пробирки, переброшенные зерна присоединяют ко второму прибору.
Добавляют по 3 мл этилового спирта, нагревают до 50° на водяной бане в течение 20 мин., вносят по 3 мл дистиллированной воды, перемешивают. По охлаждении к пробе прибавляют по 0,5 мл 4% раствора хлорамина, 0,5 мл 3% раствора фенола и по 0,5 мл 2% раствора едкого натра. Содержимое пробирок встряхивают и интенсивность окраски пробы сравнивают со шкалой стандартов (табл. 92).
Таблица 92
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
|
Рабочий стандартный раствор, мл |
0 |
0,05 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
|
Поглотительный раствор, мл |
3 |
2,95 |
2,9 |
2,8 |
2,6 |
2,4 |
2,2 |
2,0 |
|
Содержание анилина, мкг |
0 |
0,5 |
1 |
2 |
4 |
6 |
8 |
10 |
Все пробирки шкалы обрабатывают аналогично пробе.
Расчет см. выше.
При отборе проб в жидкую среду 3 мл пробы переносят в пробирку, вносят 0,3 мл 0,1 н. раствора щелочи для нейтрализации, 0,5 мл 4% раствора хлорамина, раствор встряхивают, добавляют 0,5 мл 3% раствора фенола и 0,2 мл 2% раствора щелочи. Содержимое пробирки встряхивают и через 10 — 20 мин. измеряют оптическую плотность при длине волны 660 нм в кювете с толщиной слоя 1 см по сравнению с контролем, который готовят одновременно и аналогично пробе. Содержание вещества в анализируемом объеме раствора определяют по предварительно построенному калибровочному графику. Для построения калибровочного графика готовят шкалу стандартов, но без добавления силикагеля. Все пробирки шкалы обрабатывают аналогично пробе и строят график.
Расчет см. выше.
МОНОМЕТИЛАНИЛИН
Прозрачная бесцветная жидкость, температура кипения 197,7 °C, плотность при 20 °C 0,986. Метиланилин в воде не растворим, растворим в органических растворителях.
На организм действует так же, как анилин.
Предельно допустимые концентрации: максимальная разовая и среднесуточная 0,04 мг/м3.
ОПРЕДЕЛЕНИЕ МОНОМЕТИЛАНИЛИНА С ХЛОРАМИНОМ [3]
Принцип определения
При взаимодействии метиланилина с хлорамином и фенолом в щелочной среде образуется соединение, которое окрашивает раствор в синий цвет.
Чувствительность определения — 1 мкг в анализируемом объеме раствора. Другие первичные и вторичные ароматические амины и аммиак мешают определению.
Реактивы
1. Поглотительный раствор, 2% раствор этанола.
2. Стандартный раствор метиланилина: готовят исходный стандартный раствор в мерной колбе емкостью 50 мл, в которую наливают 20 мл 40% раствора этанола, колбу взвешивают, в нее вносят 2 — 3 капли метиланилина и снова взвешивают. Раствор в колбе взбалтывают и объем его доводят до метки 40% раствором этанола. По разности двух взвешиваний устанавливают количество метиланилина в колбе и вычисляют содержание его в 1 мл раствора.
Рабочий стандартный раствор с содержанием 10 мкг/мл получают в мерной колбе емкостью 50 мл, в которую вносят такое количество исходного раствора, которое содержит 500 мкг метиланилина. Раствор доводят до метки 2% раствором этанола. Применяют свежеприготовленный раствор.
3. Этанол 96°, 2 и 40% растворы.
4. Хлорамин Б, 6% раствор. После растворения и отстаивания раствор фильтруют.
5. Фенол, 6% раствор.
6. Тиосульфат натрия, 0,1 н. раствор.
7. Едкий натр, 6% раствор.
Отбор пробы
Воздух протягивают со скоростью 0,5 л/мин. через два поглотительных прибора с пористым фильтром N 1, наполненных по 5 мл поглотительного раствора.
Ход анализа
Каждый поглотительный прибор анализируют отдельно. По 3 мл пробы из каждого поглотительного прибора переносят в пробирки, вливают по 0,5 мл хлорамина, раствор встряхивают и через 5 мин. добавляют 0,1 мл раствора тиосульфата, 0,1 мл щелочи и 0,4 мл раствора фенола. После каждого добавления растворы встряхивают. Через 20 мин. измеряют оптическую плотность раствора при длине волны 620 нм в кювете с толщиной слоя 1 см по сравнению с контролем, который готовят одновременно и аналогично пробе.
Содержание метиланилина в анализируемом объеме определяют по предварительно построенному калибровочному графику.
Для построения графика готовят шкалу стандартов (табл. 93).
Таблица 93
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
|
Рабочий стандартный раствор, мл |
0 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
2,0 |
|
Поглотительный раствор, мл |
3 |
2,9 |
2,8 |
2,6 |
2,4 |
2,2 |
2,0 |
1,0 |
|
Содержание метиланилина, мкг |
0 |
1 |
2 |
4 |
6 |
8 |
10 |
20 |
Все пробирки шкалы обрабатывают аналогично пробе.
Шкалой стандартов можно пользоваться для визуального определения, ее готовят в колориметрических пробирках одновременно с пробами.
Расчет см. выше.
ДИМЕТИЛАНИЛИН
|
C6H5N(CH3)2 |
Мол. вес 121,19 |
Бесцветная жидкость с неприятным запахом, температура кипения 193 °C, плотность при 20 °C 0,956, мало растворим в воде, но хорошо растворим в этаноле, эфире и кислотах.
Диметиланилин действует на организм подобно анилину.
Предельно допустимые концентрации: максимальная разовая, среднесуточная 0,0055 мг/м3.
ОПРЕДЕЛЕНИЕ ДИМЕТИЛАНИЛИНА С СУЛЬФАНИЛОВОЙ КИСЛОТОЙ [6]
Принцип определения
При взаимодействии диметиланилина с диазотированной сульфаниловой кислотой образуется соединение, окрашивающее раствор в розовый цвет, по интенсивности которого определяют содержание диметиланилина.
Чувствительность определения — 0,4 мкг в анализируемом объеме.
Первичные амины мешают определению.
Реактивы
1. Поглотительный раствор, 0,01 н. раствор серной кислоты.
2. Стандартный раствор диметиланилина. Готовят исходный раствор в мерной колбе емкостью 50 мл, в которую вливают 5 — 10 мл поглотительного раствора, колбу взвешивают, вносят в нее 2 — 3 капли диметиланилина и вновь взвешивают, объем жидкости в колбе доводят до метки поглотительным раствором. По разности двух взвешиваний устанавливают количество диметиланилина в колбе и рассчитывают содержание его в 1 мл раствора.
Рабочий стандартный раствор с содержанием 1 мкг/мл получают в мерной колбе емкостью 100 мл, в нее вносят такое количество исходного раствора, которое содержит 100 мкг диметиланилина. Объем жидкости в колбе доводят до метки поглотительным раствором.
3. Нитрит натрия, 0,2% раствор.
4. Едкий натр, 0,1 н. раствор.
5. Соляная кислота, 1 н. раствор.
6. Составной реактив: 0,2 г сульфаниловой кислоты растворяют в 20 мл 0,1 н. раствора едкого натра и добавляют 20 мл 0,2% раствора нитрита натрия. Объем раствора доводят до 100 мл водой. Раствор готовят перед применением.
7. Сульфаниловая кислота.
Отбор пробы
а) Для определения разовой концентрации пропускают воздух со скоростью 1 л/мин. через поглотительный прибор с пористым фильтром N 1, наполненный 3 мл поглотительного раствора. Продолжительность отбора более часа.
б) Для определения среднесуточной концентрации воздух со скоростью 0,5 л/мин. протягивают через поглотительный прибор с пористым фильтром N 1, наполненный 3 мл поглотительного раствора. Отбор проводят в течение суток. Допустимо проводить отбор 6 — 12 раз с перерывами в 2 — 4 ч в один и тот же поглотительный прибор. При наполнении прибора отмечают уровень жидкости, который периодически доводят до первоначального.
Допустимо отбор проводить 6 — 12 раз с перерывами 2 — 4 ч в один и тот же поглотительный прибор.
Ход анализа
В колориметрическую пробирку помещают 2 мл пробы. В пробирку прибавляют 2 мл составного реактива и 4 капли 1 н. раствора соляной кислоты. Раствор встряхивают и через 15 мин. интенсивность окраски пробы сравнивают со шкалой стандартов (табл. 94).
Таблица 94
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
|
Рабочий стандартный раствор, мл |
0 |
0,4 |
0,6 |
0,8 |
1,0 |
1,5 |
2,0 |
|
Поглотительный раствор, мл |
2 |
1,6 |
1,4 |
1,2 |
1,0 |
0,5 |
0 |
|
Содержание диметиланилина, мкг |
0 |
0,4 |
0,6 |
0,8 |
1,0 |
1,5 |
2,0 |
Все пробирки шкалы обрабатывают аналогично пробе и одновременно с ней.
Расчет см. выше.
МЕЗИДИН
|
(CH3)3C6H2NH |
Мол. вес 135,21 |
Маслообразная прозрачная жидкость темно-коричневого цвета с резким запахом, температура кипения 233 °C, плотность при 20 °C 0,963. Не растворим в воде, растворим в уксусной кислоте, этаноле, эфире и других органических растворителях.
Токсическое действие на организм человека сходно с действием анилина.
Предельно допустимые концентрации: максимальная разовая и среднесуточная 0,003 мг/м3.
ОПРЕДЕЛЕНИЕ МЕЗИДИНА ПО ОБРАЗОВАНИЮ ДИАЗОСОЕДИНЕНИЯ [8]
Принцип определения
При взаимодействии мезидина и нитрита натрия в кислой среде образуется диазосоединение, при сочетании последнего с ![]() -нафтолом в карбонатной среде раствор окрашивается в розово-красный цвет, по интенсивности которого определяют содержание мезидина.
-нафтолом в карбонатной среде раствор окрашивается в розово-красный цвет, по интенсивности которого определяют содержание мезидина.
Чувствительность определения — 0,1 мкг в анализируемом объеме.
Анилин мешает определению.
Реактивы
1. Поглотительный раствор 0,01 н. раствор серной кислоты.
2. Стандартный раствор мезидина: готовят исходный раствор в мерной колбе емкостью 50 мл, в которую вливают 10 — 20 мл 0,1 н. раствора серной кислоты. Колбу взвешивают, в нее вносят 2 — 3 капли мезидина и вновь взвешивают. Объем жидкости в колбе доводят до метки 0,1 н. раствором серной кислоты. По разности двух взвешиваний устанавливают содержание мезидина в колбе и вычисляют количество его в 1 мл раствора.
Рабочий стандартный раствор с содержанием 1 мкг/мл готовят соответствующим разведением исходного раствора.
3. Карбонат натрия (безводный) 5 н. раствор, свежеприготовленный.
4. Нитрит натрия, 2% раствор.
5. Серная кислота, 0,1 н. и 0,01 н. растворы.
6. ![]() -Нафтол, 5% спиртовой раствор.
-Нафтол, 5% спиртовой раствор.
Отбор пробы
а) Для определения максимально разовой концентрации воздух протягивают со скоростью 1 л/мин. через два соединенных поглотительных прибора с пористым фильтром N 1, наполненных 3 мл 0,01 н. раствором серной кислоты каждый. Продолжительность отбора 30 мин.
б) Для определения среднесуточной концентрации непрерывно в течение суток воздух со скоростью 0,5 л/мин. протягивают через указанные выше приборы. При наполнении приборов раствором отмечают уровень жидкости, который периодически добавляют водой до первоначального.
Допустимо проводить отбор с перерывами на 2 — 4 ч в те же приборы.
Ход анализа
Каждый поглотительный прибор анализируют отдельно. В колориметрическую пробирку переносят 2 мл пробы и прибавляют 0,2 мл нитрита натрия, 0,3 мл карбоната натрия и 0,2 мл раствора ![]() -нафтола. Пробирку встряхивают после каждого добавления раствора.
-нафтола. Пробирку встряхивают после каждого добавления раствора.
Через 5 мин. окрашенный раствор пробы сравнивают со шкалой стандартов, которую готовят одновременно и аналогично с пробой (табл. 95).
Расчет см. выше.
Таблица 95
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
Номер пробирки |
|||
|
1 |
2 |
3 |
4 |
||
|
Рабочий стандартный раствор, мл |
0 |
0,1 |
0,3 |
0,5 |
1,0 |
|
Поглотительный раствор, мл |
2,0 |
1,9 |
1,7 |
1,5 |
1,0 |
|
Содержание мезидина, мкг |
0 |
0,1 |
0,3 |
0,5 |
1,0 |
ПИРИДИН
Жидкость без цвета, с резким запахом. Растворим в воде, спирте, эфире, бензоле.
Плотность 0,9878 (20 °C), плотность паров по отношению к воздуху 2,75. Температура кипения 115,58 °C.
Продукты замещения атомов водорода в ядре пиридина носят название метилпроизводных; однозамещенных — пиколинов, дву- и тризамещенных — лутидинов, коллидинов. Все эти соединения относятся к пиридиновым основаниям. Пиридин и пиридиновые основания токсичны. Поражают нервную систему, вызывают раздражение слизистых оболочек дыхательных путей, глаз. Поражают печень, почки, действуют на желудочно-кишечный тракт.
Предельно допустимые концентрации: максимальная разовая и среднесуточная 0,08 мг/м3.
ОПРЕДЕЛЕНИЕ ПИРИДИНА С ХЛОРАМИНОМ И РОДАНИДОМ КАЛИЯ [5]
Принцип определения
При взаимодействии пиридина с хлорамином, роданидом калия или аммония и барбитуровой кислотой при pH 4,5 — 6,5 образуется соединение, окрашенное в розовый цвет.
Чувствительность — 0,25 мкг в определяемом объеме раствора.
Определению мешают пиридиновые основания.
Реактивы
1. Поглотительный раствор. Серная кислота 0,10 н. раствор.
2. Стандартный раствор пиридина. Чистый пиридин высушивают над прокаленной окисью кальция. Для этого в сухую склянку с притертой пробкой насыпают окись кальция и наливают пиридин. Окись кальция расплывается. Пиридин сливают в другую склянку, в которую снова вносят свежепрокаленную окись кальция. Повторяют операции до тех пор, пока окись кальция перестанет расплываться. Пиридин сливают и перегоняют. Собирают фракцию с температурой кипения 115,6 °C.
В мерную колбу емкостью 100 мл вносят около 5 мл 0,1 н. раствора серной кислоты, взвешивают. Вносят 0,1 мл перегнанного пиридина и снова взвешивают, доводят объем до метки. Получают исходный раствор с содержанием около 1 мг/мл пиридина.
Рабочий стандартный раствор с содержанием 5 мкг/мл готовят разбавлением исходного 0,1 н. раствором серной кислоты.
3. Натр едкий, 0,1 н. раствор.
4. Роданид аммония или калия, 1% раствор.
5. Хлорамин «В», 8% раствор.
6. Барбитуровая кислота, 1% раствор.
Навеску барбитуровой кислоты растворяют в воде, нагретой до 50 — 60 °C.
7. Фосфат калия 0,1 М раствор.
Навеску однозамещенного фосфата калия 13,613 г растворяют в небольшом объеме воды в мерной колбе емкостью 1 л. После растворения объем доводят до метки.
8. Фосфатнощелочной буфер, pH 6,0 — 6,2 К 50 мл 0,1 М раствора фосфата калия добавляют 5,7 мл 0,1 н. раствора едкого натра и объем доводят до 100 мл.
9. Универсальный индикатор, раствор.
Отбор пробы
а) Для определения разовой концентрации воздух со скоростью 0,2 л/мин. протягивают через два последовательно соединенных поглотительных прибора с пористой пластинкой N 1 (малая модель), содержащих по 2 мл 0,1 н. раствора серной кислоты.
Продолжительность отбора 20 — 30 мин.
б) Для определения среднесуточной концентрации воздух со скоростью 0,1 л/мин. протягивают через два последовательно соединенных поглотительных прибора с пористой пластинкой N 1 (малая модель), содержащих по 2 мл 0,1 н. раствора серной кислоты, отмечая уровень жидкости.
Отбор проводят непрерывно в течение суток, периодически добавляя воду до первоначального уровня.
Допустимо проводить отбор проб с перерывами в 2 — 4 ч в одну и ту же систему поглотителей.
Ход анализа
По 1 мл пробы переносят в пробирки.
Предварительно устанавливают количество щелочи, которое необходимо добавить в пробу для создания соответствующего pH. Для этого к 1 мл 0,1 н. раствора серной кислоты добавляют 1 мл 1% раствора роданида калия или аммония и 1 мл 8% раствора хлорамина. Через 10 мин. вносят 3 мл 1% раствора барбитуровой кислоты и 4 — 5 капель универсального индикатора и титруют 0,1 н. раствором щелочи до желтой окраски (pH 4,5 — 6,5), получают значение (а).
К пробе добавляют по 1 мл 1% раствора роданида калия или аммония, 1 мл 8% раствора хлорамина, тщательно перемешивают и через 10 мин. вносят по 3 мл 1% раствора барбитуровой кислоты и (а) мл 0,1 н. раствора щелочи, количество которой было установлено. Объем пробы доводят до объема 7 мл путем добавления фосфатнощелочного буфера.
Пробирки помещают на водяную баню и выдерживают в течение 30 — 35 мин. при 35 — 45 °C.
После охлаждения раствор фотометрируют в кюветах с толщиной слоя 1 см при длине волны 584 нм по сравнению с контролем. Содержание пиридина в анализируемом объеме определяют по предварительно построенному калибровочному графику.
Для построения калибровочного графика готовят шкалу стандартов (табл. 96).
Таблица 96
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
|
Рабочий стандартный раствор, мл |
0 |
0,05 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
|
Поглотительный раствор, мл |
1 |
0,95 |
0,9 |
0,8 |
0,6 |
0,4 |
0,2 |
0 |
|
Содержание пиридина, мкг |
0 |
0,25 |
0,5 |
1,0 |
2,0 |
3,0 |
4,0 |
5,0 |
Все пробирки шкалы обрабатывают аналогично пробе.
Шкалой стандартов можно пользоваться для визуального определения, ее готовят одновременно с пробами.
Расчет см. выше.
ДИМЕТИЛФОРМАМИД
Бесцветная жидкость со специфическим запахом, на свету не разлагается. Температура кипения 153 °C, плотность 0,96 (25°), упругость пара 3,7 мм рт. ст. Смешивается в любых отношениях с водой, спиртом, простыми и сложными эфирами. Оказывает раздражающее действие на верхние дыхательные пути, глаза и кожу. Предельно допустимые концентрации: максимальная разовая и среднесуточная 0,03 мг/м3.
ОПРЕДЕЛЕНИЕ ДИМЕТИЛФОРМАМИДА С ДИНИТРОХЛОРБЕНЗОЛОМ [2]
Принцип определения
Метод основан на гидролизе диметилформамида щелочью и взаимодействием продукта гидролиза — диметиламина с 2,4-динитрохлорбензолом. Образуется соединение, окрашенное в желтый цвет. Интенсивность окраски пропорциональна количеству диметилформамида.
Чувствительность определения — 0,5 мкг в анализируемом объеме.
Определению мешают амины.
Реактивы
1. Поглотительный раствор — соляная кислота, 0,05 н. раствор.
2. Стандартные растворы диметилформамида. Исходный стандартный раствор готовят следующим образом. В мерную колбу емкостью 25 — 50 мл вливают 10 — 15 мл 0,05 н. раствора соляной кислоты, взвешивают, вносят 2 — 3 капли диметилформамида и снова взвешивают. Объем жидкости в колбе доводят до метки 0,05 н. раствором соляной кислоты и хорошо перемешивают. По разности в весе определяют количество диметилформамида и вычисляют его содержание в 1 мл раствора. Рабочий стандартный раствор с содержанием 10 мкг/мл диметилформамида готовят соответствующим разведением исходного раствора 0,05 н. раствором соляной кислоты.
3. Соляная кислота, 0,01 н., 0,05 н. и 25% растворы.
4. 20% раствор едкого натра или едкого кали.
5. Карбонат натрия кристаллический, 8% раствор.
6. 2,4-Динитрохлорбензол, 1% спиртовой раствор. Готовят при нагревании на водяной бане.
7. Хлороформ.
Отбор пробы
а) Для определения разовой концентрации воздух со скоростью 1 л/мин. протягивают через поглотительный прибор Зайцева, содержащий 5 мл 0,05 н. раствора соляной кислоты. Продолжительность отбора 20 — 30 мин.
б) Для определения среднесуточной концентрации воздух со скоростью 0,25 л/мин. протягивают через поглотительный прибор Зайцева, содержащий 5 мл 0,05 н. раствора соляной кислоты. Отбор проводят непрерывно в течение суток, периодически добавляя воду до первоначального уровня. Допустимо отбор проводить 6 — 12 раз с перерывами в 2 — 4 ч в одни и те же поглотительные приборы.
Ход анализа
В поглотительный прибор с пробой для гидролиза диметилформамида вливают 1 мл 20% раствора едкого натра. Этот поглотительный прибор соединяют с другим поглотительным прибором Зайцева, наполненным 4 мл 0,01 н. раствора соляной кислоты. Поглотительный прибор с пробой помещают в кипящую водяную баню точно на 5 мин., баня должна быть закрытой, и в это время через поглотительные приборы пропускают воздух со скоростью 0,1 л/мин. Образовавшийся диметиламин поглощается 0,01 н. раствором соляной кислоты. Для исключения возможности переброса жидкости поглотительный прибор с продуктом гидролиза быстро отъединяют от всей системы при включенном аспираторе. Для анализа берут в колориметрическую пробирку 2 мл раствора. Одновременно готовят шкалу стандартов, для этого в поглотительные приборы Зайцева через длинную трубку вносят рабочий стандартный раствор в количестве 0,1; 0,2; 0,8; 1,2; 1,6; 2 мл и доводят до 4 мл 0,05 н. раствором соляной кислоты. Кислоту вливают также через длинную трубку поглотителя, чтобы смыть с ее стенок диметилформамид. Во все поглотительные приборы вливают по 1 мл 20% раствора щелочи и обрабатывают так же, как и пробу. Для составления шкалы стандартов после гидролиза берут в колориметрические пробирки половину, т.е. 2 мл раствора, что соответствует следующим количествам диметилформамида (табл. 97).
Таблица 97
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
|
Содержание диметилформамида, мкг |
0 |
0,5 |
1 |
2 |
4 |
6 |
8 |
10 |
Контрольный опыт ставят так же, как и пробу. Во все пробирки шкалы и проб прибавляют по 0,2 мл 8% раствора карбоната натрия, растворы встряхивают и к ним добавляют по 0,2 мл 1% раствора динитрохлорбензола. Все пробирки помещают на 5 мин. в кипящую водяную баню. После охлаждения в пробирки прибавляют по 0,2 мл 25% раствора соляной кислоты и после взбалтывания растворов добавляют по 0,5 мл хлороформа. Растворы снова встряхивают и через 15 — 20 мин. сравнивают желтую окраску проб со шкалой стандартов.
Расчет см. выше.
ЛИТЕРАТУРА
1. Алексеева М.В. Определение анилина в воздухе. В сб.: Предельно допустимые концентрации атмосферных загрязнений. М., «Медгиз», 1963, в. 7.
2. Алексеева М.В. Определение диметилформамида. В кн.: Определение атмосферных загрязнений. М., «Медицина», 1963.
3. Беляков А.А., Горбылева Н.В. Определение микрограммовых количеств диметиланилина, анилина и метиланилина в смеси. Аналитическая химия, 1957, т. 12, 4.
4. Виноградова В.А., Помазова Е.Н. Определение низших алифатических аминов в воздухе. Всесоюзная конференция по новым методам определения микроконцентраций токсических веществ в воздухе и других средах. Харьков, 1972.
5. Горская Р.В., Ярым-Агаева Н.Т. Фотометрическое определение малых количеств пиридина. Аналитическая химия, 1965, 6.
6. Зильберг Л.А. К вопросу об определении диметиланилина в воздухе производственных помещений. Гиг. и сан., 1957, 6.
7. Кузнецов В.И., Пименова З.М. и др. Определение гексаметилендиамина. В кн.: Определение вредных веществ в воздухе. М., Медгиз, 1957.
8. Полежаев Н.Г., Кузнецова Л.В. Новый колориметрический метод определения мезидина в воздухе. Гиг. и сан., 1967, 4.
Глава XIV
РАЗНЫЕ ОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
КАПРОЛАКТАМ
Бесцветное кристаллическое вещество, растворимое в воде и органических растворителях, температура плавления 68 — 70 °C, температура кипения 139 — 140 °C (12 мм рт. ст.).
Капролактам вызывает головные боли, раздражительность, нарушение сна, при действии на кожу может вызывать дерматиты.
Предельно допустимые концентрации капролактама (пары, аэрозоль): максимальная разовая и среднесуточная 0,06 мг/м3.
ОПРЕДЕЛЕНИЕ КАПРОЛАКТАМА С ГИДРОКСИЛАМИНОМ [7]
Принцип определения
При воздействии капролактама с гидроксиламином в щелочной среде образуется гидроксамовая кислота, которую определяют колориметрически по реакции с хлорным железом.
Чувствительность определения — 50 мкг капролактама в анализируемом объеме раствора.
Определению мешают сложные эфиры карбоновых кислот и другие вещества, реагирующие с гидроксиламином с образованием гидроксамовой кислоты.
Определению не мешают циклогексанон, циклогексанол, циклогексаноноксим, нитроциклогексан, бензол, аммиак, сернистый газ и серный ангидрид.
Реактивы
1. Поглотительный раствор — дистиллированная вода.
2. Стандартный раствор капролактама. Исходный стандартный раствор, содержащий 1000 мкг/мл капролактама, готовят путем растворения 0,1 г капролактама в 100 мл воды.
3. Гидроксиламин солянокислый, 3 М раствор.
В 100 мл воды растворяют 21 г солянокислого гидроксиламина. Раствор должен быть свежеприготовленный.
4. Хлорид железа, 10% раствор. Растворяют 10 г хлорида окисного железа в 80 мл воды и добавляют 15 мл 2 н. раствора соляной кислоты. Раствор годен к употреблению в течение недели.
5. Натр едкий, 1 н. раствор.
6. Соляная кислота, 2 н. раствор.
7. Контрольный раствор. К 10 мл воды добавляют 4 мл раствора гидроксиламина и 1 мл раствора едкого натра. Контрольный раствор готовят перед употреблением.
Отбор пробы
а) Для определения максимальной разовой концентрации исследуемый воздух протягивают со скоростью 10 л/мин. через фильтр АФА-ХС, укрепленный в патроне, и через поглотительный прибор Гернет, содержащий 8 мл дистиллированной воды, соединенный последовательно с патроном.
б) Для определения среднесуточной концентрации воздух со скоростью 2 — 5 л/мин. непрерывно в течение суток протягивают через фильтр АФА-ХС, укрепленный в патроне, и поглотительный прибор Гернет, содержащий 8 мл дистиллированной воды, отмечая уровень жидкости и периодически добавляя воду до первоначального объема. Допустимо отбор проводить на один и тот же фильтр и поглотитель с перерывами в 2 или 4 ч.
Ход анализа
Фильтр переносят в стакан, промывают 5 мл воды, отжимая стеклянной палочкой. Раствор сливают в круглодонную колбу емкостью 75 — 100 мл, добавляют 2 мл раствора солянокислого гидроксиламина и 0,5 мл 1 н. раствора едкого натра. Затем присоединяют обратный холодильник, раствор нагревают и кипятят точно 30 мин. от начала закипания. Раствор охлаждают до комнатной температуры, сливают в пробирку, колбу ополаскивают 0,5 мл контрольного раствора, который сливают в ту же пробирку.
Для анализа берут в колориметрическую пробирку 3 мл пробы. Из поглотительного прибора Гернет берут 5 мл пробы и обрабатывают, как описано выше.
Для составления шкалы стандартов проводят гидролиз капролактама для перевода его в гидроксамовую кислоту. В день анализа в колбу с обратным холодильником вносят 10 мл рабочего стандартного раствора, добавляют 4 мл гидроксиламина, 1 мл раствора едкого натра, кипятят точно 30 мин. от начала закипания, охлаждают. Доводят объем до 15 мл и получают рабочий стандартный раствор с содержанием 667 мкг/мл капролактама. Рабочий стандартный раствор наносят на фильтры, применяемые при отборе, и готовят шкалу стандартов (табл. 98).
Таблица 98
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
|
Раствор гидроксамовой кислоты, полученной после гидролиза капролактама, мл |
0 |
0,075 |
0,12 |
0,15 |
0,23 |
0,30 |
|
Содержание капролактама, мкг |
0 |
50 |
75 |
100 |
150 |
200 |
Контрольный фильтр и фильтры с нанесенными стандартными растворами помещают в стакан и обрабатывают аналогично пробам. Одновременно во все пробирки шкалы и пробы добавляют по 0,1 мл раствора 2 н. соляной кислоты и по 0,5 мл 10% раствора хлорного железа. Через 10 мин. сравнивают интенсивность окраски проб со шкалой стандартов. Окраска шкалы стандартов от зеленовато-желтой до красновато-коричневой, устойчива в течение 30 мин.
По шкале стандартов (см. табл. 98) колориметрируют пробы, отобранные на фильтры АФА-Х-11, определение же количества капролактама, поглощенного в воду в поглотителях Гернет, производят по другой стандартной шкале (табл. 99), приготовленной из стандартного раствора, полученного также после гидролиза капролактама.
Таблица 99
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
|
Раствор гидроксамовой кислоты, полученной после гидролиза капролактама, мл |
0 |
0,08 |
0,12 |
0,15 |
0,23 |
0,30 |
|
Контрольный раствор, мл |
3 |
2,92 |
2,88 |
2,85 |
2,77 |
2,70 |
|
Содержание капролактама, мкг |
0 |
50 |
75 |
100 |
150 |
200 |
Одновременно во все пробирки шкалы и пробы добавляют по 0,1 мл 2 н. раствора соляной кислоты, 0,5 мл 10% раствора хлорного железа, перемешивают и через 10 мин. колориметрируют.
Расчет см. выше.
Примечание. Количество капролактама, обнаруженное в смыве с фильтра и в пробе из поглотителя Гернет, суммируют.
ОКИСЬ ЭТИЛЕНА
|
|
Мол. вес 44,03 |
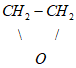
Бесцветный газ, при низких температурах — жидкость с эфирным запахом; удельный вес 0,8713 (20 °C), температура кипения 10,7 °C, упругость пара 1,095 мм рт. ст. (20 °C). Растворим в воде, эфире и спирте. Окись этилена способна к реакции со многими органическими и неорганическими веществами, полимеризируется, при нагревании с водой образует этиленгликоль, при 400 °C получается изомерный ацетальдегид.
Обладает наркотическим действием и сильным общетоксическим действием на организм.
Предельно допустимые концентрации окиси этилена: максимальная разовая и среднесуточная 0,03 мг/м3.
ОПРЕДЕЛЕНИЕ ОКИСИ ЭТИЛЕНА ПО ФОРМАЛЬДЕГИДУ [3]
Принцип определения
При поглощении окиси этилена водным раствором серной кислоты образуется этиленгликоль, последний окисляют йодной кислотой до формальдегида. Формальдегид определяют колориметрически с хромотроповой кислотой. Чувствительность определения 0,5 мкг в анализируемом объеме раствора. Определению мешают формальдегид, этиленгликоль.
Реактивы
1. Поглотительный раствор: 40% раствор серной кислоты.
2. Стандартный раствор окиси этилена. Готовят исходный раствор, для чего берут мерную колбу емкостью 50 мл, наливают в нее 10 — 15 мл 40% раствора серной кислоты, взвешивают, добавляют в колбу 2 — 3 капли окиси этилена и снова взвешивают. Раствор в колбе доводят до метки 40% раствором серной кислоты и тщательно перемешивают.
Рассчитывают содержание окиси этилена в 1 мл раствора и готовят рабочие стандартные растворы А и Б с содержанием 100 мкг/мл (раствор А) и 10 мкг/мл (раствор Б).
Стандартные растворы можно готовить из этиленгликоля.
Раствор А должен содержать 140 мкг/мл этиленгликоля, рабочий стандартный раствор — 14 мкг/мл, что соответствует 100 и 10 мкг окиси этилена.
3. Перйодат калия (KIO4), 3% раствор в 10% растворе серной кислоты. Растворение проводят при нагревании.
4. Сульфит натрия, 50% раствор. Раствор устойчив в течение 2 — 3 сут. Раствор готовят при нагревании.
5. Хромотроповая кислота. 0,15 г реактива растворяют в 5 мл воды, затем прибавляют 125 мл концентрированной серной кислоты. Раствор устойчив в течение 2 — 3 дней.
6. Серная кислота, концентрированная, 10 и 40% растворы.
Отбор пробы
а) Для определения разовой концентрации воздух со скоростью 0,5 л/мин. протягивают через два поглотительных прибора с пористой пластинкой N 1, содержащих по 4 мл поглотительного раствора.
Продолжительность отбора 20 — 30 мин.
б) Для определения среднесуточной концентрации воздух со скоростью 0,25 — 0,3 л/мин. протягивают через два поглотительных прибора с пористой пластинкой N 1, содержащих по 4 мл поглотительного раствора, отмечая уровень жидкости. Отбор проводят непрерывно в течение суток, периодически добавляя воду до первоначального уровня. Допустимо проводить отбор 6 — 12 раз с перерывом в 2 — 4 ч в одни и те же поглотительные приборы.
Ход анализа
В колориметрическую пробирку из каждого поглотительного прибора вносят 3 мл пробы, приливают по 0,5 мл 3% раствора перйодата калия, перемешивают и оставляют на 30 мин. Далее добавляют по 3 капли раствора сульфита натрия для восстановления избытка перйодата калия, по 1,5 мл хромотроповой кислоты, перемешивают и погружают на 30 мин. в кипящую водяную баню. По охлаждении добавляют 2 мл воды и перемешивают. При этом коричневый фон, обусловленный выделением йода, исчезает и остается фиолетовое окрашивание, характерное для реакции формальдегида с хромотроповой кислотой. После охлаждения растворы переливают в кюветы с толщиной слоя 1 см и фотометрируют при длине волны 574 нм по сравнению с контролем, который готовят одновременно и аналогично пробам. Содержание окиси этилена в анализируемом объеме определяют по предварительно построенному калибровочному графику.
Для построения калибровочного графика готовят шкалу стандартов (табл. 100).
Таблица 100
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
|
Рабочий стандартный раствор Б, мл |
0 |
0,05 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
2,0 |
|
Поглотительный раствор, мл |
3 |
2,95 |
2,9 |
2,8 |
2,6 |
2,4 |
2,2 |
2,0 |
1,0 |
|
Содержание окиси этилена, мкг |
0 |
0,5 |
1,0 |
2,0 |
4,0 |
6,0 |
8,0 |
10,0 |
20,0 |
Все пробирки шкалы обрабатывают аналогично пробам, фотометрируют и строят калибровочный график.
Шкалой стандартов можно пользоваться и для визуального определения, ее готовят в колориметрических пробирках одновременно с пробами.
Примечание. Очистку хромотроповой кислоты см. выше.
Расчет см. выше.
ТОЛУИЛЕНДИИЗОЦИАНАТ
|
(CH3)C6H3(NCO)2 |
Мол. вес 174,0 |
Прозрачная слегка желтоватая жидкость с неприятным запахом. Толуилендиизоцианат известен в двух изомерах: 2,4- и 2,6-толуилендиизоцианат. Перегоняется под вакуумом, в воде гидролизуется, хорошо растворяется в органических растворителях. Смесь изомеров 2,4- и 2,6-толуилендиизоцианатов в отношении 65:35 обладает температурой кипения 162 °C при 5 мм рт. ст.
Толуилендиизоцианат действует раздражающе на слизистые оболочки верхних дыхательных путей и глаз.
Предельно допустимые концентрации: максимальная разовая 0,05 мг/м3 и среднесуточная 0,02 мг/м3.
ОПРЕДЕЛЕНИЕ ТОЛУИЛЕНДИИЗОЦИАНАТА С ![]() -НАФТОЛОМ [8]
-НАФТОЛОМ [8]
Принцип определения
При взаимодействии диамина, образующегося при омылении толуилендиизоционата щелочью, с нитритом натрия в солянокислой среде происходит диазотирование. При сочетании диазосоединения с ![]() -нафтолом образуется соединение, окрашивающее раствор в оранжево-красный цвет.
-нафтолом образуется соединение, окрашивающее раствор в оранжево-красный цвет.
Чувствительность определения — 0,5 мкг в анализируемом объеме раствора.
Толуилендиамин, мешающий определению, задерживают на фильтре АФА.
Реактивы
1. Поглотительный раствор — 0,1 н. раствор едкого натра.
2. Стандартный раствор толуилендиизоцианата. Готовят исходный раствор в мерной колбе емкостью 50 мл, в которую наливают 15 — 20 мл ацетона, колбу взвешивают, в нее вносят 2 — 3 капли толуилендиизоцианата и снова взвешивают. Раствор в колбе доводят до метки ацетоном и хорошо перемешивают. По разности двух взвешиваний устанавливают количество толуилендиизоцианата в колбе и высчитывают его содержание в 1 мл раствора.
Рабочий стандартный раствор с содержанием 10 мкг/мл получают в мерной колбе емкостью 50 мл, в нее вносят такое количество исходного раствора, в котором содержится 500 мкг толуилендиизоцианата. Раствор в колбе доводят до метки поглотительным раствором.
3. Нитрит-бромидный реактив. 0,5 г бромистого натрия растворяют в 100 мл 0,3% раствора нитрита натрия.
4. ![]() -Нафтол, 0,1% раствор в 1 н. растворе едкого натра.
-Нафтол, 0,1% раствор в 1 н. растворе едкого натра.
Готовят за 10 мин. до применения.
5. Натр едкий, 1 н. раствор.
6. Соляная кислота, 0,2 н. раствор.
7. Нитрит натрия, 0,3% раствор.
Отбор пробы
а) Для определения максимальной разовой концентрации воздух со скоростью 0,5 л/мин. протягивают через поглотительный прибор с пористым фильтром N 1, наполненный 5 мл поглотительного раствора. Продолжительность отбора 20 — 30 мин.
б) Для определения среднесуточной концентрации воздух со скоростью 0,3 л/мин. протягивают через поглотительный прибор, наполненный 5 мл поглотительного раствора. Если в процессе отбора пробы происходит изменение объема, то периодически в поглотительный прибор вносят воду и доводят объем до первоначального уровня. Допустимо отбор пробы проводить 6 — 12 раз с перерывами в 2 — 4 ч в один и тот же поглотительный прибор.
Ход анализа
В пробирку переносят 2,5 мл пробы, добавляют 2,8 мл 0,2 н. раствора соляной кислоты. Пробу помещают в сосуд со льдом на 5 мин. После этого в пробирку вносят 0,2 мл нитрит-бромидного реактива, содержимое перемешивают и добавляют 0,2 мл ![]() -нафтола. Раствор тщательно перемешивают и через 10 мин. измеряют оптическую плотность в кювете с толщиной слоя 1 см при длине волны 496 нм по сравнению с контролем.
-нафтола. Раствор тщательно перемешивают и через 10 мин. измеряют оптическую плотность в кювете с толщиной слоя 1 см при длине волны 496 нм по сравнению с контролем.
Контроль готовят одновременно и аналогично пробе.
Содержание толуилендиизоцианата в анализируемом объеме определяют по заранее построенному калибровочному графику. Для построения графика готовят шкалу стандартов (табл. 101).
Таблица 101
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
|
Рабочий стандартный раствор, мл |
0 |
0,05 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
|
|
Поглотительный раствор, мл |
2,5 |
2,45 |
2,4 |
2,3 |
2,1 |
1,9 |
1,7 |
1,5 |
|
|
Содержание толуилендиизоцианата, мкг |
0 |
0,5 |
1 |
2 |
4 |
6 |
8 |
10 |
Все пробирки шкалы обрабатывают аналогично пробе. Шкалой стандартов можно пользоваться для визуального определения, ее готовят в колориметрических пробирках одновременно с пробами.
Расчет см. выше.
ТИОФЕН
(Тиофуран)
|
|
Мол. вес 84,14 |
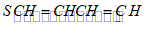
Бесцветная жидкость с температурой кипения 84 °C, температурой плавления — 38,3 °C. Плотность 1,064 (20,1 °C). Тиофен не растворим в воде, растворим в спирте, эфире, бензоле; устойчив к действию окислителей, легко реагирует с ртутью, дает металлоорганические соединения.
Пары тиофена вызывают раздражение слизистых оболочек дыхательных путей. Обладает наркотическим действием.
Предельно допустимая максимальная разовая концентрация тиофена 0,6 мг/м3.
СПЕКТРОФОТОМЕТРИЧЕСКОЕ ОПРЕДЕЛЕНИЕ ТИОФЕНА [1]
Принцип определения
Метод основан на измерении оптической плотности спиртового раствора тиофена при длине волны 231 нм.
Чувствительность определения — 2,5 мкг в анализируемом объеме раствора. Определению мешают вещества, поглощающие при длине волны 231 нм.
Реактивы
1. Поглотительный раствор — метиловый спирт, перегнанный при 64 — 65 °C.
2. Стандартные растворы тиофена. В мерную колбу емкостью 25 мл с притертой пробкой наливают 10 — 15 мл метилового спирта и взвешивают на аналитических весах, затем вносят 2 — 3 капли тиофена и снова взвешивают. Объем доводят метиловым спиртом до метки, тщательно перемешивают и по разности между первым и вторым взвешиванием рассчитывают содержание тиофена в 1 мл. Соответствующим разведением готовят стандартный раствор с содержанием 0,1 мг/мл, а из него разведением в 10 раз готовят рабочий стандартный раствор с содержанием 10 мкг/мл.
3. Тиофен, х.ч.
Отбор пробы
Для определения разовой концентрации воздух со скоростью 0,1 — 0,2 л/мин. протягивают через 2 — 3 поглотительных прибора с пористой пластинкой N 1, содержащих 6 мл метилового спирта. Во время отбора пробы поглотительный прибор помещают в сосуд со льдом. Продолжительность отбора 20 — 30 мин.
Ход анализа
Содержимое каждого поглотительного прибора переливают в кварцевую кювету с толщиной слоя 1 см и фотометрируют на спектрофотометре при длине волны 231 нм. В качестве контроля применяют метиловый спирт. Содержание тиофена в анализируемом объеме определяют по предварительно построенному калибровочному графику. Для построения калибровочного графика готовят шкалу стандартов (табл. 102).
Таблица 102
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
|
Рабочий стандартный раствор, мл |
0 |
0,25 |
0,5 |
1 |
2 |
3 |
4 |
5 |
|
Метиловый спирт, мл |
5 |
4,75 |
4,5 |
4,0 |
3,0 |
2,0 |
1,0 |
0 |
|
Содержание тиофена, мкг |
0 |
2,5 |
5,0 |
10,0 |
20,0 |
30,0 |
40,0 |
50,0 |
Растворы шкалы стандартов фотометрируют в тех же условиях, что и пробы.
Расчет см. выше.
ДИВИНИЛ
(1,3-Бутадиен)
|
CH2=CH-CH=CH2 |
Мол. вес 54,09 |
Газ с неприятным запахом, не растворимый в воде и растворимый в органических растворителях. Температура кипения 4,5 °C, температура плавления -109 °C, легко полимеризируется.
Дивинил оказывает раздражающее действие на слизистые оболочки верхних дыхательных путей, глаз; может вызывать дерматиты, в высоких концентрациях действует как наркотик.
Предельно допустимые концентрации дивинила: максимальная разовая 3 мг/м3, среднесуточная 1 мг/м3.
СПЕКТРОФОТОМЕТРИЧЕСКИЙ МЕТОД ОПРЕДЕЛЕНИЯ
ДИВИНИЛА В ПРИСУТСТВИИ ![]() -МЕТИЛСТИРОЛА
-МЕТИЛСТИРОЛА
И ИЗОПРОПИЛБЕНЗОЛА [4]
Принцип определения
Количественное определение дивинила проводят по спектрам поглощения изооктанового раствора трехкомпонентной системы дивинила, изопропилбензола и альфа-метилстирола при длинах волн 224 и 245 нм.
Чувствительность определения — 0,1 мкг/мл раствора. Определению мешают вещества, поглощающие в рабочих длинах волн 224 и 245 нм.
Реактивы
1. Силикагель марки КСМ и МСМ с зернением 1 мм, предварительно обработанный.
2. Изооктан эталонный, оптически чистый. Для получения оптически чистого изооктана эталонный изооктан дополнительно очищается с помощью колонки, заполненной силикагелем, до получения растворителя с оптической плотностью ~ 0,175 относительно дистиллированной воды при 220 нм в кювете с толщиной слоя 1 см.
3. Стеклянная вата.
Отбор пробы
Для определения разовой концентрации воздух со скоростью 1 л/мин. протягивают через V-образную стеклянную трубку диаметром 4 мм, заполненную 0,5 г силикагеля. Трубка закрыта тампонами из стеклянной ваты и стеклянными заглушками. При отборе пробы заглушки снимают. При наличии запаха дивинила достаточно отобрать 1 л воздуха, при отсутствии запаха следует отобрать 5 — 10 л воздуха.
Ход анализа
Силикагель из V-образной трубки переносят в пробирку с притертой пробкой, заливают 4 мл изооктана, тщательно перемешивают, закрывают пробкой и оставляют в горизонтальном положении при частом встряхивании на 1 ч. Прозрачную жидкость над слоем силикагеля сливают, помещают в кварцевую кювету с толщиной слоя 1 см и измеряют оптическую плотность на спектрофотометре. Как эталон сравнения ставят изооктан, полученный путем обработки чистого силикагеля.
При длине волны 224 нм находят суммарную оптическую плотность (E1), при длине волны 245 нм находят оптическую плотность (E2) мешающей примеси.
Расчет анализа
Концентрацию дивинила (C) в микрограммах на 1 мл вычисляют по формуле:
C = 2,5 (E — 0,47 E2).
Концентрацию дивинила в миллиграммах на 1 м3 воздуха рассчитывают по обычной формуле (см. выше).
Примечание. Коэффициенты 0,47 и 2,5 постоянные. 0,47 — постоянное соотношение оптической плотности ![]() -метилстирола при 224 и 245 нм. Оптическая плотность дивинила с концентрацией 1 мкг/мл при 224 нм составляет 1 / 4 = 2,5.
-метилстирола при 224 и 245 нм. Оптическая плотность дивинила с концентрацией 1 мкг/мл при 224 нм составляет 1 / 4 = 2,5.
ОПРЕДЕЛЕНИЕ ОБЩЕГО КОЛИЧЕСТВА ОЗОНА И ОКСИДАНТОВ
С РОДАНИСТЫМ АММОНИЕМ [2]
Принцип определения
Метод основан на реакции взаимодействия оксидантов с солью Мора в кислой среде с образованием ионов трехвалентного железа, которое определяется колориметрически в виде железороданидного комплекса.
Чувствительность определения по озону — 0,3 мкг в анализируемом объеме раствора. Определению не мешают сероводород, формальдегид, акролеин, углеводороды. Определению мешает сернистый газ в концентрациях, превышающих оксиданты в 50 — 100 раз. Двуокись азота входит в состав оксидантов.
Реактивы
1. Поглотительный раствор. 0,1 г соли Мора растворяют в 100 мл дистиллированной воды, добавляют 10 мл 1 М азотной кислоты и 10 мл ацетона. Поглотительный раствор должен быть свежеприготовленный и стоять на холоду.
2. Стандартный раствор перекиси водорода. Для приготовления стандартного раствора 5 — 10 мл 30% раствора перекиси водорода растворяют в 500 мл дистиллированной воды в мерной колбе. Для титрования в коническую колбу берут 10 мл раствора, прибавляют 200 мл воды, 20 мл серной кислоты (1:3) и титруют 0,1 н. раствором перманганата калия до появления розового окрашивания. 1 мл 0,1 н. раствора перманганата калия соответствует 1,7 мг перекиси водорода. Путем соответствующего разбавления готовят раствор с содержанием 10 мкг/мл и рабочий стандартный раствор с содержанием 1 мкг/мл. Стандартные растворы должны быть свежеприготовленными.
3. Азотная кислота, 1 М раствор. Перед приготовлением 1 М раствора концентрированную азотную кислоту продувают током воздуха с целью удаления окислов азота.
4. Соль Мора FeSO4·(NH4)2SO4·6H2O, перекристаллизованная.
5. Ацетон.
6. Роданистый аммоний. Реактив должен быть бесцветным или слегка розоватым.
7. Роданистый аммоний, 30% раствор. Раствор должен быть бесцветным или слегка розоватым. При хранении на холоду устойчив в течение нескольких дней.
8. Серная кислота 1:3 (по объему).
9. Перманганат калия, 0,1 н. раствор.
Отбор пробы
Исследуемый воздух со скоростью 0,5 л/мин. протягивают через два поглотительных прибора, наполненных по 5 мл поглотительного раствора.
Ход анализа
Из каждого поглотительного прибора переносят 5 мл пробы в колориметрические пробирки и приливают по 2 мл 30% раствора роданистого аммония. Через 5 — 10 мин. фотометрируют образующуюся окраску в кюветах с толщиной слоя 1 см при длине волны 480 — 490 нм по сравнению с контролем, который готовят одновременно и аналогично пробе. Содержание оксидантов в анализируемом объеме раствора определяют по предварительно построенному градуировочному графику. Для построения градуировочного графика готовят шкалу стандартов (табл. 103).
Таблица 103
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
|
Рабочий стандартный раствор, мл |
0 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
2,0 |
|
Поглотительный раствор, мл |
5 |
4,8 |
4,6 |
4,4 |
4,2 |
4,0 |
3,0 |
|
Содержание перекиси водорода, мкг |
0 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
2,0 |
|
Эквивалентное содержание озона, мкг |
0 |
0,28 |
0,57 |
0,85 |
1,13 |
1,41 |
2,82 |
Шкалу стандартов обрабатывают аналогично пробе, измеряют оптическую плотность растворов и строят график. Шкалу стандартов можно использовать для визуального определения. В этом случае ее строят одновременно с пробами. Окраска устойчива в течение 30 — 40 мин.
Расчет анализа см. выше.
Примечание. Поглотительные приборы с пористой пластинкой или поглотительный прибор Зайцева перед отбором пробы воздуха должны быть отмыты от следов железа. Для этого предварительно хорошо вымытые поглотительные приборы заливают поглотительным раствором на 4 — 5 ч; раствор выливают, поглотители промывают еще 2 — 3 раза поглотительным раствором с помощью резинового баллона, раствор сливают, добавляют 2 мл роданистого аммония и измеряют его оптическую плотность. Оптическая плотность раствора должна быть меньше 0,01. Отмытые поглотительные приборы хранят залитыми дистиллированной водой и закрытыми заглушками.
ИНГИБИТОРЫ АТМОСФЕРНОЙ КОРРОЗИИ
Ингибиторами называются вещества, тормозящие химические реакции. Они защищают от атмосферной коррозии черные и цветные металлы.
НИТРИТ ДИЦИКЛОГЕКСИЛАМИНА (НДА)
|
|
Мол. вес 228,1 |
Твердое вещество белого цвета. Температура плавления 180 °C, упругость пара 0,1 · 103 мм рт. ст. Плохо растворимо в воде, растворимо в этаноле, метаноле, ацетоне. Токсично. Предельно допустимая концентрация: максимальная разовая 0,02 мг/м3.
ОПРЕДЕЛЕНИЕ НИТРИТА ДИЦИКЛОГЕКСИЛАМИНА (НДА)
С РЕАКТИВОМ ГРИССА-ИЛОСВАЯ [9]
Принцип определения
При взаимодействии НДА с реактивом Грисса-Илосвая образуется азокраситель, окрашивающий раствор в розовый цвет.
Чувствительность определения — 0,5 мкг НДА в анализируемом объеме раствора. Определению мешает двуокись азота.
Реактивы
1. Поглотительный раствор — дистиллированная вода.
2. Стандартный раствор НДА. Исходный стандартный раствор с содержанием 100 мкг/мл готовят растворением 0,01 г НДА в 100 мл дистиллированной воды. Рабочий стандартный раствор с содержанием 10,0 мкг/мл готовят соответствующим разведением исходного раствора.
Остальные реактивы — см. «Определение двуокиси азота».
Отбор пробы
Для определения разовой концентрации воздух со скоростью 2 — 3 л/мин. протягивают через фильтр АФА, помещенный в патрон, соединенный с двумя поглотительными приборами Рыхтера, наполненными по 6 мл воды.
Ход анализа
В пробирки вносят по 5 мл пробы из каждого поглотительного прибора. Фильтр извлекают из патрона, переносят в стакан и заливают 3 мл воды, отжимают на стенке стакана стеклянной палочкой и снова обрабатывают 3 мл воды. В пробирку помещают 5 мл раствора. Во все пробирки добавляют по 0,5 мл реактива Грисса-Илосвая, перемешивают, оставляют стоять 20 — 30 мин. и фотометрируют при длине волны 530 нм в кювете с толщиной слоя 1 см по сравнению с контролем, который готовят одновременно и аналогично пробе. Содержание НДА в анализируемом объеме раствора определяют по предварительно построенному калибровочному графику. Для построения графика готовят шкалу стандартов (табл. 104).
Таблица 104
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
|
Рабочий стандартный раствор, мл |
0 |
0,05 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
|
Поглотительный раствор, мл |
5 |
4,95 |
4,9 |
4,8 |
4,6 |
4,4 |
4,2 |
4,0 |
|
Содержание НДА, мкг |
0 |
0,5 |
1 |
2 |
4 |
6 |
8 |
10 |
Все пробирки шкалы обрабатывают аналогично пробе, измеряют оптическую плотность и строят график.
Расчет см. выше.
МЕТАНИТРОБЕНЗОАТГЕКСАМЕТИЛЕНИМИН (Г-2) [9]
|
|
Мол. вес 226,0 |
Твердое вещество белого цвета. Температура плавления 133 °C, упругость пара 1 · 106 мм рт. ст. Плохо растворим в воде, растворим в этаноле. Относится к токсическим соединениям.
Предельно допустимая концентрация: максимальная разовая 0,02 мг/м3.
ОПРЕДЕЛЕНИЕ МЕТАНИТРОБЕНЗОАТАГЕКСАМЕТИЛЕНИМИНА
(ИНГИБИТОРА — Г-2) С n-ДИМЕТИЛАМИНОБЕНЗАЛЬДЕГИДОМ [9]
Принцип определения
Метод основан на восстановлении метанитробензотагексаметиленимина с последующим определением по реакции конденсации с парадеметиламинобензальдегидом.
Чувствительность определения — 3 мкг в анализируемом объеме раствора.
Определению мешают некоторые первичные ароматические амины.
Реактивы
1. Стандартные растворы Г-2.
Стандартный раствор Г-2 готовят растворением 0,01 г ингибитора в 100 мл дистиллированной воды. Содержание Г-2 в растворе 100 мкг/мл.
2. Парадиметиламинобензальдегид, 5% раствор в 5% растворе соляной кислоты.
3. Соляная кислота, концентрированная.
4. Соляная кислота, 5% раствор.
5. Гранулированный цинк.
Отбор пробы
Для определения разовой концентрации исследуемый воздух со скоростью 3 л/мин. протягивают через фильтр АФА-В, помещенный в патрон, соединенный с двумя поглотительными приборами Рыхтера, наполненными по 6 мл воды. Продолжительность отбора 20 — 30 мин.
Ход анализа
В пробирки вносят 3 мл пробы из каждого поглотительного прибора, а фильтр извлекают и переносят в стакан. Фильтр смачивают 1 — 2 каплями этилового спирта, заливают 3 мл воды, промывают и отжимают стеклянной палочкой на воронке. Операцию повторяют.
В колориметрические пробирки переносят 3 мл жидкости. Во все пробы добавляют по 0,5 мл концентрированной соляной кислоты и по одной грануле цинка. Восстановление проводят при 100 — 110 °C на песчаной бане в течение 5 мин. После охлаждения пробы отфильтровывают в колориметрические пробирки, приливают 1,5 мл 5% раствора парадиметиламинобензальдегида в 5% растворе соляной кислоты, перемешивают, оставляют стоять в течение 10 мин. и фотометрируют в кювете с толщиной слоя 1 см при длине волны 445 нм по сравнению с контролем, который готовят одновременно и аналогично пробе.
Содержание Г-2 в анализируемом объеме определяют по предварительно построенному градуировочному графику, который строят по шкале стандартов (табл. 105).
Таблица 105
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
0 |
1 |
2 |
3 |
4 |
5 |
6 |
|
Стандартный раствор, мл |
0 |
0,03 |
0,05 |
0,1 |
0,2 |
0,4 |
0,8 |
|
Поглотительный раствор, мл |
3 |
2,97 |
2,95 |
2,9 |
2,8 |
2,6 |
2,2 |
|
Содержание Г-2, мкг |
0 |
3 |
5 |
10 |
20 |
40 |
80 |
Шкалу стандартов обрабатывают аналогично пробам и строят график. Определение можно проводить и визуально.
Расчет анализа см. выше.
МАСЛОРАСТВОРИМАЯ СОЛЬ ДИЦИКЛОГЕКСИЛАМИНА (МСДА)
(C6H11)2HChH2h-1COOH
Маслорастворимая соль дициклогексиламина представляет собой бурое, пастообразное вещество с температурой плавления 350 °C, не растворимое в воде, растворимое в спирте (3 г на 100 г), ацетоне и бензоле. Обладает токсическим действием. Предельно допустимая концентрация: максимальная разовая 0,008 мг/м3.
ОПРЕДЕЛЕНИЕ МАСЛОРАСТВОРИМОЙ СОЛИ ДИЦИКЛОГЕКСИЛАМИНА (МСДА)
С ДИЭТИЛДИТИОКАРБАМАТОМ НАТРИЯ [9]
Принцип определения
Метод основан на образовании медных солей жирных кислот с последующим определением их по реакции с диэтилдитиокарбаматом натрия.
Чувствительность определения — 5 мкг в анализируемом объеме раствора. Определению мешают жирные кислоты.
Реактивы
1. МСДА.
2. Стандартные растворы МСДА в хлороформе.
Для приготовления исходного стандартного раствора с содержанием 1 мг/мл в бюксе взвешивают 25 мг. МСДА растворяют в хлороформе, переносят в мерную колбу емкостью 25 мл и доливают хлороформом до метки.
Из исходного стандартного раствора путем соответствующего разбавления готовят рабочий стандартный раствор, содержащий 100 мкг/мл.
3. Хлороформ, х.ч.
4. Триэтаноламин, х.ч. 1 М раствор.
5. Уксусная кислота, ледяная.
6. Уксусная кислота, 1 М раствор.
7. Медь сернокислая, 6,5% водно-спиртовой раствор.
8. Составной раствор готовят из 9 частей 1 М триэтаноламина, 1 части 1 М уксусной кислоты и 10 частей 6,5% раствора сернокислой меди (раствор применяется свежеприготовленный).
9. Бутиловый спирт.
10. Диэтилдитиокарбамат натрия, 0,1% раствор в бутиловом спирте (раствор должен быть свежеприготовленный).
11. Беззольные фильтры «синяя лента».
Отбор пробы
Воздух со скоростью 3 — 10 л/мин. протягивают через бумажный фильтр, помещенный в патрон.
Ход анализа
Фильтр с отобранной пробой помещают в коническую колбу, заливают 6 мл хлороформа. Фильтр промывают в хлороформе, отжимают стеклянной палочкой на воронке и добавляют 5 мл составного раствора. Колбу помещают в аппарат для встряхивания на 1 1/2 — 2 мин.
Содержимое колбы переносят в делительную воронку, разделяют смесь и сливают хлороформный слой через воронку с беззольным фильтром, предварительно смоченным хлороформом, в пробирку с притертой пробкой. К 5 мл хлороформного экстракта добавляют 0,5 мл 0,1% раствора диэтилдитиокарбамата натрия в бутиловом спирте, перемешивают и фотометрируют в кювете с толщиной слоя 1 см при длине волны 440 нм по сравнению с контролем, который готовят одновременно и аналогично пробе.
Содержание МСДА в анализируемом объеме определяют по предварительно построенному калибровочному графику.
Для построения калибровочного графика готовят шкалу стандартов, при этом рабочий стандартный раствор наносят на фильтры (табл. 106).
Таблица 106
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
|
Рабочий стандартный раствор, мл |
0 |
0,05 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
|
Содержание МСДА, мкг |
0 |
5 |
10 |
20 |
40 |
60 |
80 |
100 |
Одновременно готовят контрольный фильтр. Контрольный фильтр и фильтры, содержащие различное количество МСДА, помещают в колбы с притертой пробкой и далее обрабатывают аналогично пробе, измеряют оптическую плотность растворов и строят график.
Расчет см. выше.
1,4-НАФТОХИНОН
|
|
Мол. вес 158,15 |
Ярко-желтые кристаллы с температурой плавления 125°, легко возгоняющиеся. 1,4-Нафтохинон не растворяется в воде, растворяется в органических растворителях, окислитель.
Применяется в производстве синтетических красителей. Токсичен, обладает резко выраженным раздражающим свойством, действует на нервную систему, почки.
2,3-ДИХЛОР-1,4-НАФТОХИНОН
|
|
Мол. вес 227,0 |
Твердое кристаллическое вещество желтого цвета с температурой плавления 193 °C, растворимое в горячем спирте, не растворимое в воде. Применяется в химической промышленности. Токсичен, обладает резорбтивным и местным раздражающим действием на теплокровных животных.
Предельно допустимые концентрации: максимальная разовая 0,05 мг/м3, среднесуточная 0,05 мг/м3.
СПЕКТРОФОТОМЕТРИЧЕСКОЕ ОПРЕДЕЛЕНИЕ 1,4-НАФТОХИНОНА
И 2,3-ДИХЛОР-1,4-НАФТОХИНОНА [10]
Принцип определения
Метод основан на способности 2,3-дихлор-1,4-нафтохинона поглощать свет при длине волны 280 нм, тогда как 1,4-нафтохинон в этой области прозрачен. В то же время эти соединения имеют интенсивную полосу поглощения при длине 246 нм. Использование метода Фирордта позволяет по величине оптической плотности смеси двух компонентов определять концентрацию каждого из них.
Чувствительность определения — 1 мкг/мл.
Реактивы
1. Этанол.
2. Фильтры АФА-ХА-18.
Отбор пробы
Воздух протягивают со скоростью 2 л/мин. через фильтр АФА, закрепленный в патроне, последовательно соединенный с двумя поглотительными приборами, содержащими по 10 мл этанола.
Отбор проб проводят при охлаждении.
Ход анализа
Фильтр переносят в стакан и вливают 10 мл этанола. В течение 10 мин. помешивают стеклянной палочкой. Фильтр осторожно отжимают и промывают его 5 мл этанола. Экстракты соединяют, сюда же переносят и содержимое поглотительных приборов. Измеряют объем пробы.
Раствор (4 мл) переносят в кювету с толщиной слоя 1 см и измеряют оптические плотности при 246 и 280 нм. Измерения проводят по отношению к этанолу.
Предварительно из стандартных этанольных растворов, в которых содержится по 100 мкг/мл 1,4-нафтохинона или 2,3-дихлор-1,4-нафтохинона, готовят стандартную шкалу. В каждой пробирке шкалы должно содержаться по 1 — 3 — 5 — 7 — 10 мкг/мл раствора того или другого вещества. Измеряют оптические плотности растворов 2,3-дихлор-1,4-нафтохинона и отдельно 1,4-нафтохинона при 246 нм.
Кроме того, измеряют оптические плотности растворов 2,3-дихлор-1,4-нафтохинона при длине волны 280 нм.
Строят три градуировочных графика:
1) для 2,3-дихлор-1,4-нафтохинона при 246 нм,
2) для 1,4-нафтохинона при 246 нм,
3) для 2,3-дихлор-1,4-нафтохинона при 280 нм.
По градуировочному графику (3) находят концентрацию 2,3-дихлор-1,4-нафтохинона на основании полученной величины оптической плотности при измерении пробы.
Затем по градуировочному графику (1) находят величину оптической плотности, соответствующую найденной концентрации 2,3-дихлор-1,4-нафтохинона. Разность оптических плотностей указывает на величину оптической плотности, соответствующую 1,4-нафтохинону. Концентрацию 1,4-нафтохинона определяют по калибровочному графику (2). Таким образом находят концентрации того и другого соединения в 1 мл исследуемого раствора. При расчете учитывают объем всей пробы.
Расчет см. выше.
БЕНЗ(А)ПИРЕН <1>
———————————
<1> По старой номенклатуре — 3,4-бензпирен
|
|
Мол. вес 252,3 |
Бенз(а)пирен (БП) твердое кристаллическое вещество, температура плавления 179 °C, стойкое к внешним воздействиям.
БП относится к пятикольчатым ароматическим углеводородам, обладает высокой канцерогенной активностью.
Источниками образования и выделения БП в атмосферный воздух и другие внешние среды являются процессы термической переработки органического сырья, сжигания топлив и др.
Среднесуточная предельно допустимая концентрация 0,1 мкг на 100 м3 воздуха. При изучении уровня загрязнения атмосферного воздуха бенз(а)пиреном применяют аспирационный и седиментационный способы отбора проб. При аспирационном способе исследуемый воздух протягивают через фильтры ФПП-15, ФПА-15 со скоростью до 250 м3/ч в течение от 20 мин. до нескольких часов. Обычно накапливают до 4 г пыли, в которой определяют количество БП. В ряде случаев отбор проб проводят в поглотительные приборы, наполненные бензолом, со скоростью 0,5 — 1 л/мин. в течение 20 — 50 мин.
При седиментационном способе отбирают снеговые пробы или собирают пыль в специальные сосуды.
Из анализируемой пробы извлекают фракции, содержащие БП, очищают их от сопутствующих примесей хроматографированием.
Количественное определение БП проводят методом квазилинейчатых спектров люминесценции (эффект Шпольского), позволяющим устанавливать БП в концентрациях 10-8 — 10-10 г/мл (0,01 — 0,0001 мкг/мл).
Подготовка проб к анализу, специальная обработка посуды, растворителей, а также проведение анализа подробно изложены в двух нормативных документах, утвержденных Министерством здравоохранения СССР в 1972 г. [5, 6].
ЛИТЕРАТУРА
1. Аигина Е.Л., Лопухова А.И. и др. Определение тиофена в воздухе спектрофотометрическим методом. Гиг. и сан., 1966, 6.
2. Дмитриев М.Т., Соловьева Т.В. и др. Определение оксидантов. Гиг. и сан., 1972, 2.
3. Крылова Н.А. Определение окиси этилена в атмосферном воздухе, Гиг. и сан., 1961, 10.
4. Манита М.Д. Спектрофотометрический метод определения дивинила в присутствии альфа-метилстирола и изопропилбензола. Гиг. и сан., 1965, 8.
5. Методические указания по отбору проб из объектов внешней среды и их подготовке к анализу на канцерогенные полициклические ароматические углеводороды Министерства здравоохранения СССР. М., 1972, N 930-71.
6. Методические указания по определению канцерогенных полициклических ароматических углеводородов, в частности, бенз(а)пирена, в различных промышленных и природных продуктах Министерства здравоохранения СССР. М., 1972, N 931-71.
7. Русских А.А. Колориметрический метод определения капролактама в воздухе. Гиг. труда, 1965, 9.
8. Семенов В.А., Нехорошева Е.В. и др. Модификация колориметрического метода 2,4-толуилендиизоцианата с ![]() -нафтолом. Гиг. и сан., 1971, 4.
-нафтолом. Гиг. и сан., 1971, 4.
9. Соловьева Т.В., Евсеенко Н.С. и др. Определение ингибиторов атмосферной коррозии. Гиг. и сан., 1972, 1.
10. Черниченко Н.А. Спектрофотометрическое определение 1,4-нафтохинона в воздухе. Инструкция Киевского института общей и коммунальной гигиены им. А.Н.
Глава XV
САЖА, ПЫЛЬ, ДВУОКИСЬ КРЕМНИЯ В ПЫЛИ
САЖА
Сажа — продукт неполного сгорания или термического разложения углеродистых веществ. В различных видах сажи содержится углерода от 88,5 до 99,9%, водорода от 0,1 до 1%, кислорода от 0,01 до 10% и минеральных веществ от 0,01 до 0,6%. Сажа — высокодисперсный порошок, размер частиц ее от 10 до 600 ммк, удельный вес 1,6 — 2 г/см3. Сажа не растворяется ни в кислотах, ни в щелочах. Сажа применяется в полиграфической, лакокрасочной, резиновой промышленности. В производстве сажи, при большой запыленности, отмечены заболевания верхних дыхательных путей, органов дыхания и пищеварения, наблюдаются раздражения роговицы глаз и конъюнктивы. Некоторые виды сажи (например, полученные из пека) могут вызывать образование на коже злокачественных опухолей. Увеличение заболеваемости раком легких иногда объясняют наличием в воздухе сажи, в которой найдено до 0,03% 3,4-бензпирена. Сажа значительно уменьшает прозрачность атмосферы, что приводит к снижению освещенности, видимости и ультрафиолетовой радиации.
Предельно допустимые концентрации сажи (копоти): максимальная разовая 0,15 мг/м3, среднесуточная 0,05 мг/м3.
ОПРЕДЕЛЕНИЕ САЖИ. МЕТОД 1 [5]
Принцип определения
Сажу улавливают на бумажный фильтр. Степень почернения фильтра сопоставляют со шкалой, приготовленной из суспензии чистой сажи, нанесенной в определенных количествах на фильтровальную бумагу.
Чувствительность определения — 2 мкг. Мешают вещества, окрашивающие фильтр.
Аппаратура и реактивы
1. Патрон для отбора проб, внутренний диаметр 5 мм.
2. Беззольные фильтры («синяя лента»).
3. Вазелиновое масло.
4. Сапонин.
5. Стандартная шкала для определения сажи. Ее приготовляют следующим образом. Навеску сажи (около 120 мг), полученную путем сжигания чистого вазелинового масла, тщательно растирают в фарфоровой ступке с 2 — 3 мл дистиллированной воды, к которой прибавляют крупинку сапонина. После этого полученную смесь переносят в колбу емкостью 1 л, доводят водой до метки и тщательно взбалтывают. При этом получается равномерная и хорошо стабилизированная суспензия. При наличии на поверхности суспензии отдельных плавающих конгломератов сажи их осторожно снимают прикосновением фильтровальной бумаги.
Часть суспензии (500 мл) выпаривают в доведенной до постоянного веса фарфоровой чашке и снова высушивают до постоянного веса. Определяют истинную концентрацию сажи в стандартной суспензии. Она оказывается близкой к 0,1 мг/мл. Из этой основной суспензии разведением в мерной колбе приготовляют более слабую, с содержанием 10 мкг/мл.
Определенные объемы основной суспензии или ее десятикратного разведения фильтруют через бумажный фильтр, зажатый в описанном выше патроне. После фильтрования патрон промывают несколько раз водой, развинчивают, фильтр извлекают и высушивают при комнатной температуре. Фильтровальная бумага должна быть достаточно плотной и не пропускать частички сажи, что проверяют пробной фильтрацией суспензии через двойной фильтр. В этих условиях на втором листке фильтра не должно быть заметно почернения. Получающуюся серию фильтров наклеивают клейстером из рисового крахмала на ватманскую бумагу, покрывают тонким прозрачным стеклом и окантовывают по краям. Шкала может служить неопределенно долгое время.
Всего в шкале делается 10 ступеней: 2; 4; 6; 8; 10; 15; 20; 30; 40 и 50 мкг сажи.
Отбор пробы
а) Для определения разовой концентрации пробы отбирают с подветренной стороны от дымящего объекта. Воздух протягивают через бумажный фильтр, укрепленный в патроне, в течение 20 — 30 мин. со скоростью 1 л/мин.
б) Для определения среднесуточной концентрации воздух протягивают непрерывно в течение суток через такой же фильтр. Допустимо вместо круглосуточной аспирации отбирать пробу через один и тот же фильтр с перерывом в 2 — 4 ч. Общий объем протянутого воздуха должен составить 240 л; скорость протягивания воздуха 1 л/мин.
Ход анализа
Извлеченный из патрона фильтр сопоставляют по окраске со шкалой и подбирают наиболее близкую к окраске фильтра ступень шкалы.
Расчет анализа см. выше.
ОПРЕДЕЛЕНИЕ САЖИ. МЕТОД 2 [3]
Принцип определения
Сажу вместе с пылью улавливают на фильтре АФА-В. При обработке фильтра дихлорэтаном материал фильтра (перхлорвинил) и органические вещества пыли растворяются в дихлорэтане, основная высокодисперсная углеродистая составная часть сажи переходит во взвешенное суспендированное состояние, а минеральная часть пыли оседает на дно. Содержание сажи в суспензии определяют или визуально по почернению мембранного фильтра при помощи сопоставления со стандартной шкалой, или фотометрически.
Чувствительность определения — 2 мкг.
Методом можно определить визуально 0,5 мкг, фотометрически 3 мкг сажи.
Аппаратура и реактивы
1. Мембранные фильтры N 5.
2. Вазелиновое масло.
3. Дихлорэтан (хранить в темной склянке).
4. Сапонин.
5. Основная суспензия сажи.
Навеску сажи около 20 мг получают путем сжигания чистого вазелинового масла, тщательно растирают в фарфоровой ступке с 3 — 4 мл дихлорэтана, к которому прибавляют крупинку сапонина. Смесь переносят в колбу емкостью 100 мл, доводят дихлорэтаном до метки и тщательно взбалтывают. При этом получают равномерную и хорошо стабилизированную суспензию. Если на поверхности суспензии имеются отдельно плавающие конгломераты сажи, то их осторожно снимают прикосновением фильтровальной бумаги. В бюксе выпаривают 30 мл суспензии и доводят до постоянного веса. В основной суспензии сажи должно содержаться около 200 мкг/мл.
6. Стандартную суспензию сажи с содержанием 10 мкг/мл готовят из основной суспензии соответствующим разведением дихлорэтаном в мерной колбе.
Приготовление стандартной шкалы
Для приготовления стандартной шкалы определенные объемы стандартной суспензии сажи фильтруют через мембранные фильтры N 5, зажатые в патроне и колбе для отсасывания.
После фильтрования патрон каждый раз промывают дихлорэтаном и фильтры извлекают. Описанным способом приготовляют шкалу, отвечающую следующим количествам сажи: 0,5; 1; 2; 4; 7; 10; 15; 20 и 40 мкг. Серию фильтров наклеивают клейстером из рисового крахмала на бумагу и покрывают стеклом. Шкала устойчива длительное время.
Отбор пробы
Исследуемый воздух в количестве не менее 400 л протягивают через фильтр АФА-В-18, укрепленный в патроне, со скоростью 50 — 60 л/мин.
Ход анализа
а) Визуальное определение
Фильтр осторожно извлекают из патрона пинцетом и, если нужно, определяют содержание пыли. Опрессованные края фильтра обрезают и вкладывают в центрифужную пробирку с притертой пробкой. В пробирку вливают 3 мл дихлорэтана, закрывают пробкой и взбалтывают до полного растворения фильтра и получения однородной суспензии. При этом материал фильтра (перхлорвинил) и органические вещества пыли растворяются в дихлорэтане, основная высокодисперсная углеродистая составная часть сажи переходит во взвешенное суспендированное состояние, а минеральная часть пыли оседает на дно пробирки. Через 2 — 5 мин. суспензию из пробирки декантируют в пробирку с притертой пробкой и меткой 5 мл. В центрифужную пробирку вливают еще 2 мл дихлорэтана и также обрабатывают содержимое пробирки. Через 2 — 5 мин. остаточную суспензию декантируют в ту же пробирку. Из пробирки пипеткой берут 0,5 или 1 мл суспензии в зависимости от количества сажи и вносят на мембранный фильтр N 5, укрепленный в патроне и колбе для отсасывания, суспензию фильтруют. После фильтрования фильтр вынимают из патрона и содержание сажи определяют визуально, сопоставляя почернение фильтра со стандартной шкалой.
б) Фотометрическое определение
Фильтр обрабатывают в два приема 5 — 10 мл дихлорэтана, как указано выше (см. «Визуальное определение»). Через 2 — 5 мин. суспензию декантируют в пробирку с притертой пробкой. После перемешивания всю суспензию в количестве 5 мл выливают в кювету с толщиной слоя 1 см и фотометрируют с сине-фиолетовым светофильтром с максимумом пропускания при 400 нм. Количество сажи в пробе определяют по калибровочному графику.
Построение калибровочного графика
Для построения калибровочного графика в пробирки с меткой 5 мл помещают чистые фильтры АФА-В с обрезанными опрессованными краями. На фильтры наносят стандартную суспензию сажи в количествах 0,3; 0,6; 1; 1,5; 2; 3; 4 и 5 мл и доводят дихлорэтаном до объема 5 мл. Приготовленные растворы соответствуют 3; 6; 10; 15; 20; 30; 40 и 50 мкг сажи в 5 мл раствора. Содержимое пробирок взбалтывают до полного растворения фильтра, выливают в кюветы и фотометрируют. Найденные величины оптической плотности помещают в графике напротив величин содержания сажи (мкг) в анализируемом объеме. Контрольную пробу готовят из чистого фильтра АФА-В-18 с обрезанными опрессованными краями, растворенного в 5 мл дихлорэтана.
Расчет анализа см. выше.
ПЫЛЬ
Пыль относится к твердым атмосферным загрязнениям. Пыль, находящаяся в атмосферном воздухе, может быть как природного происхождения, так и результатом промышленной и бытовой деятельности человека. Концентрация взвешенных частиц является показателем загрязнения атмосферного воздуха.
В атмосферном воздухе населенных мест установлены предельно допустимые концентрации для пыли нетоксичной: максимальная разовая 0,5 мг/м3, среднесуточная 0,15 мг/м3.
ОПРЕДЕЛЕНИЕ ПЫЛИ [1]
Принцип определения
Воздух протягивают через фильтр из ткани ФПП-15-1,5. Концентрацию пыли определяют по привесу.
Чувствительность определения — 0,3 мг.
Аппаратура
Автомобильный аспиратор системы Института имени Ф.Ф. Эрисмана (рис. 28). Он состоит из Т-образной трубы (3), патрона-насадки (2), манометра (4) и разборного конусовидного стакана (1).
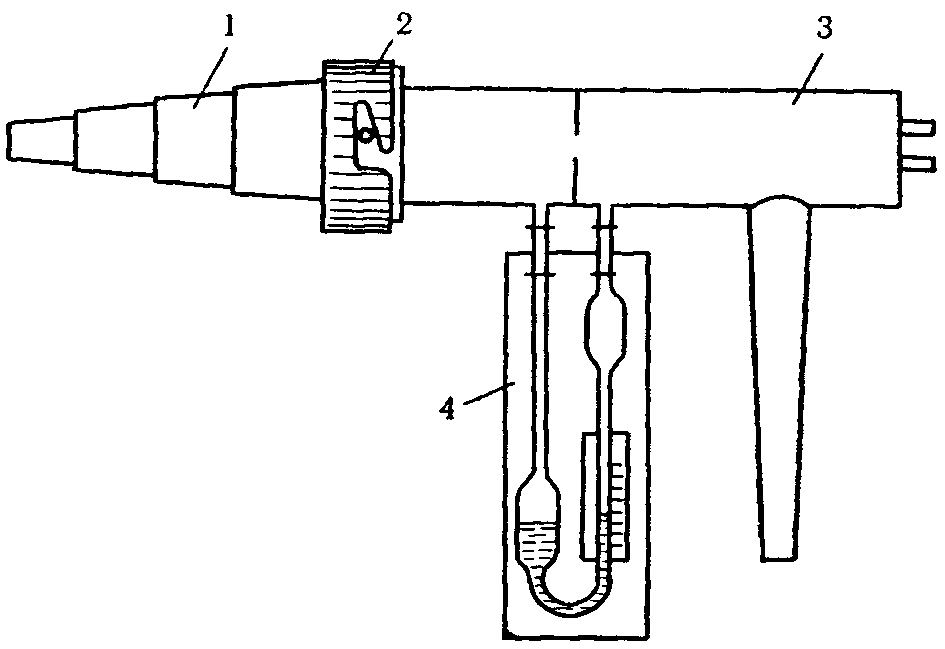
Рис. 28. Общий вид автомобильного аспиратора
а) Т-образная труба из разборного конусовидного стакана. Один конец трубы имеет втулку для укрепления патрона-насадки и два боковых отростка, к которым присоединяется манометр типа реометра, служащий для установления скорости отбора проб. Между этими отростками в трубе помещена диафрагма. Второй, противоположный, конец трубы имеет два отверстия, закрывающиеся дырчатой заслонкой, служащей для впуска воздуха в трубу мимо патрона насадки. Третий конец трубы (более узкий) служит для присоединения аспиратора с входным отверстием карбюратора автомашины.
б) Патрон-насадка (рис. 29), состоящий из корпуса, кассеты, крышки.
Корпус служит для крепления патрона в Т-образной трубке — аспирационной части автомобильного аспиратора. В корпус помещают кассету с фильтром, с наружной стороны его имеются два штифта, с помощью которых крепится крышка-насадка.
Кассета состоит из двух частей: собственно кассеты, дном которой является сетка, применяемая в аллонжах, и разрезного кольца, служащего для крепления фильтра. Кольцо по краям разреза снабжено двумя отверстиями диаметром по 2 мм, в которые вставляют пинцет для того, чтобы извлечь кольцо из кассеты. При обследовании рекомендуется иметь запас кассет (20 — 30) и в лабораторных условиях их заряжать взвешенными фильтрами.
Крышка служит для крепления кассеты с фильтром в патроне-насадке, а также для присоединения конусовидного стакана к патрону. С внутренней стороны крышка снабжена прокладкой для полноты герметичности крепления кассеты. На верхней стенке крышки имеется бортик, на который надевают разборный конусовидный стакан.
Рис 29. Патрон-насадка в разобранном виде.
1 — крышка; 2 — кассета; 3 — корпус
Разборный конусовидный стакан состоит из шести конусов, вставленных один в другой, диаметр их 11,7; 20; 25; 32; 40; 56 мм. Конусовидный стакан является средством уравнивания скорости отбора проб и скорости движения воздуха, а также для обеспечения поступления воздуха на всю рабочую поверхность фильтра, исключив возможность образования турбулентных завихрений, для чего угол корпуса не должен превышать 15°.
Для установления диаметра отверстия конуса измеряют скорость движения воздуха с помощью представленного графика (рис. 30), в котором на оси абсцисс отложена скорость ветра в метрах в секунду, а на оси ординат — соответствующие диаметры входных отверстий стаканов.
Установление диаметра стакана производится следующим образом: из точки на оси абсцисс, которая соответствует движению скорости ветра, например, 5 м/с, проводят перпендикуляр до пересечения с кривой для отбора проб со скоростью, например, 150 л/мин. Точку пересечения соединяют с осью ординат линией, параллельной оси абсцисс, и в точке соприкосновения этой линии с осью ординат будет искомый диаметр входного отверстия конусовидного стакана, в данном случае — 25 мм. Если необходимый диаметр не соответствует входному отверстию имеющихся шести диаметров стаканов, берут стакан с наиболее близким диаметром.
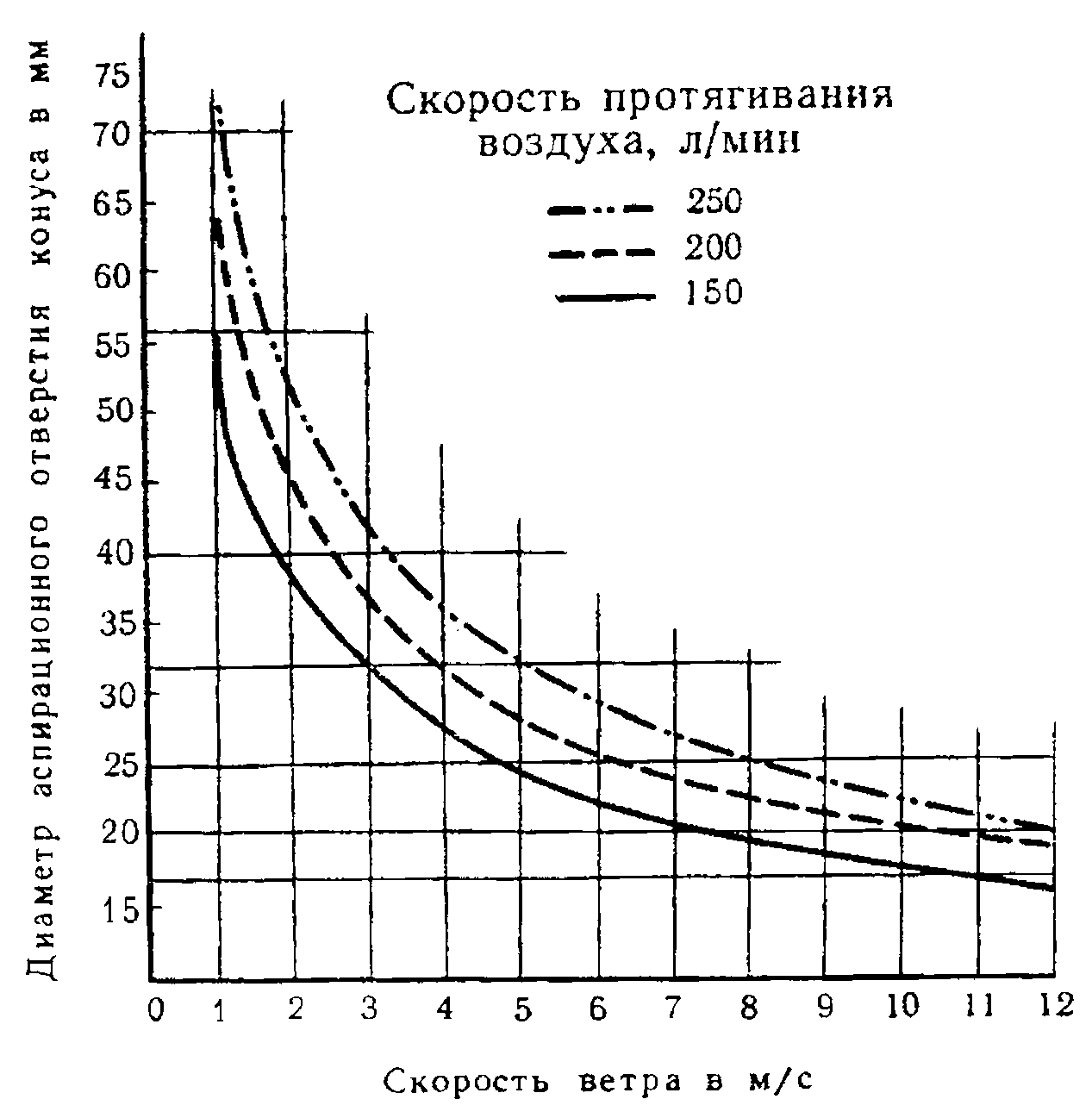
Рис. 30. График зависимости диаметра входных отверстий
конуса от скорости ветра
в) Манометр типа реометра. Шкала тарирована на производительность аспиратора в литрах в минуту. Заполняется керосином.
Отбор пробы
а) Для определения разовой концентрации воздух протягивают через фильтр из ткани ФПП-15-1,5 в течение 15 — 20 мин. со скоростью 150 — 200 л/мин., т.е. 3 м3.
б) При определении среднесуточной концентрации воздух протягивают через указанный выше фильтр 12 раз по 10 мин. со скоростью 100 л/мин. через равные интервалы.
В качестве побудителя отсоса воздуха могут быть использованы бытовые пылесосы типа «Ракета», «Уралец» и др., работающие от сети. При этом пылесос обеспечивает отбор одновременно двух параллельных проб воздуха. При работе с автомобильным аспиратором необходимо соблюдать следующую последовательность действий:
1. Разобрать патрон-насадку поворотом против часовой стрелки и снять крышку.
2. Извлечь кольцо из кассеты.
3. Вынуть фильтр из пакета.
4. Уложить фильтр на сетку кассеты с помощью пинцета и расправить края фильтра.
5. Вставить кольцо в кассету пинцетом.
6. Надеть крышку патрона-насадки и повернуть ее по часовой стрелке до упора.
7. Вставить патрон-насадку в Т-образную трубу автомобильного аспиратора.
8. Присоединить конусовидный стакан к патрону-насадке с соответствующим диаметром входного отверстия.
9. Включить аспиратор и, пользуясь дырчатой заслонкой, помещенной на противоположном конце, где находится патрон-насадка, отрегулировать протягивание воздуха по манометру.
10. Направить отверстие конуса патрона-насадки на факел выброса.
11. Отметить время начала отбора пробы.
12. После отбора пробы снять крышку патрона-насадки, пинцетом вынуть кольцо кассеты, на сетке сложить фильтр вчетверо рабочей поверхностью внутрь и уложить фильтр в пакет из кальки.
Ход анализа
В лаборатории пробы пыли помещают в весовую комнату и через 40 — 60 мин. производят взвешивание. Если отбор проб производится при большой влажности (туман или мелкий дождь), то фильтры после отбора выдерживают в эксикаторе над хлоридом кальция в течение 2 — 3 ч до постоянного веса.
Расчет анализа см. выше.
ДВУОКИСЬ КРЕМНИЯ
Двуокись кремния (кварц) представляет собой бесцветное вещество с температурой плавления 1713° и плотностью 2,62 — 2,65.
Свободная двуокись кремния может быть в кристаллической и аморфной формах. К кристаллическим минералам группы свободной двуокиси кремния относятся кварц, тридимит, кристоболит и др.; к аморфным — опал, трепел, диатомит и др. В воде и кислотах двуокись кремния не растворима, только плавиковая кислота растворяет двуокись кремния по реакции:

При сплавлении двуокиси кремния со щелочами или карбонатами образуют соли кремниевой кислоты — силикаты. При вдыхании пыли, в которой содержится кристаллическая двуокись кремния, силикаты, могут развиться такие заболевания, как силикоз, силикатоз, которые характеризуются разрастанием соединительной ткани в органах дыхания и другими изменениями.
СУММАРНОЕ ОПРЕДЕЛЕНИЕ ДВУОКИСИ КРЕМНИЯ
И СИЛИКАТОВ В ПЫЛИ [2]
Принцип определения
Метод основан на сплавлении соединений кремния с карбонатом калия — натрия и колориметрическом определении кремния в растворе по синему кремнемолибденовому комплексу.
Чувствительность определения — 1 мкг двуокиси кремния в анализируемом объеме раствора. Соединения фосфора и мышьяка не мешают определению.
Для проведения анализа требуются платиновый тигель с крышкой, тигельные щипцы с платиновыми наконечниками, агатовая ступка, платиновая или алюминиевая палочка, тигельная или муфельная печь.
Реактивы
1. Двуокись кремния SiO2 в виде чистого кварца.
2. Стандартный раствор двуокиси кремния 0,02 г тонкоизмельченной SiO2 тщательно смешивают с 0,2 г KNaCO3 и сплавляют, как указано в ходе анализа. Плав растворяют в горячей воде и раствор переносят в мерную колбу емкостью 200 мл. По охлаждении объем раствора доводят водой до метки.
Раствор, содержащий 0,1 мг/мл SiO2, сохраняют в парафинированной или полиэтиленовой склянке. Для приготовления рабочего стандартного раствора с содержанием 10 мкг/мл исходный раствор разбавляют в 10 раз водой непосредственно перед употреблением.
Стандартный раствор можно приготовить из силиката натрия Na2SiO3·9H2O (х.ч.). 0,0473 г силиката растворяют в мерной колбе в 100 мл воды. В растворе содержится 0,1 мг SiO2 в 1 мл и он сохраняется в парафинированной или полиэтиленовой склянке. Соответствующим разбавлением водой готовят раствор, содержащий 10 мкг/мл SiO2.
3. Серная кислота, 10 н. и 1 н. растворы.
4. Винная кислота, 5% раствор.
5. Аскорбиновая кислота, 1% раствор.
6. Карбонат калия — натрия KNaCO3, безводный и измельченный. Можно применить смесь безводных K2CO3 и Na2CO3, взятых в соотношении 5:4.
Отбор пробы
Исследуемый воздух со скоростью до 100 л/мин. протягивают через фильтр АФА или бумажный беззольный фильтр, помещенный в патрон <1>.
———————————
<1> Для определения можно использовать пробы пыли, причем навеска должна быть не менее 20 мг.
Ход анализа
Фильтр осторожно вынимают из патрона воронки, складывают пылью внутрь и помещают в платиновый тигель. Перед проведением анализа тигель очищают сплавлением в нем небольших количеств KNaCO3.
Тигель помещают в слегка нагретую печь, где фильтр постепенно обугливается, затем по мере повышения температуры печи его сжигают до золы. В тигель по охлаждении вносят 0,1 г карбоната натрия — калия (KNaCO3) и тщательно перемешивают с помощью алюминиевой палочки. Тигель закрывают крышкой, помещают в печь, нагретую до 800 — 900°, где смесь выдерживают до получения однородной расплавленной массы. Затем тигель вынимают из печи и по охлаждении сплав выщелачивают небольшими порциями воды. Раствор нагревают в тигле почти до кипения и сливают в мерную колбу емкостью 25 мл.
Раствор сплава осторожно подкисляют 1 н. раствором серной кислоты до слабокислой реакции на лакмус (кислотность раствора не должна быть выше 0,02 н.) и доводят водой до метки.
В колориметрическую пробирку вносят исследуемого раствора 4 мл, добавляют при встряхивании 0,1 мл раствора молибдата аммония. Через 5 мин. наливают 1 мл раствора винной кислоты и 0,1 мл раствора аскорбиновой кислоты. Спустя 20 мин. фотометрируют в кюветах с толщиной слоя 1 — 2 см при длине волны 600 нм по сравнению с контролем, который готовят одновременно и аналогично пробам. Содержание двуокиси кремния в анализируемом объеме определяют по предварительно построенному калибровочному графику. Для построения калибровочного графика готовят шкалу стандартов (табл. 107).
Таблица 107
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
9 |
10 |
11 |
|
Рабочий стандартный раствор, мл |
0 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
1,2 |
1,4 |
1,6 |
1,8 |
2,0 |
|
Вода, мл |
4 |
3,9 |
3,8 |
3,6 |
3,4 |
3,2 |
3,0 |
2,8 |
2,6 |
2,4 |
2,2 |
2,0 |
|
Содержание двуокиси кремния, мкг |
0 |
1 |
2 |
4 |
6 |
8 |
10 |
12 |
14 |
16 |
18 |
20 |
Все пробирки шкалы обрабатывают аналогично пробам, фотометрируют и строят калибровочный график. Шкалой стандартов можно пользоваться для визуального определения, ее готовят в колориметрических пробирках одновременно с пробами.
Расчет см. выше.
ОПРЕДЕЛЕНИЕ СВОБОДНОЙ ДВУОКИСИ КРЕМНИЯ В ПЫЛИ [4]
Принцип определения
Метод основан на сплавлении свободной двуокиси кремния и силикатов с бикарбонатом калия или натрия в присутствии хлоридов. При этом свободная двуокись кремния переходит в растворимую щелочную соль кремниевой кислоты, а кремниевая кислота силиката сохраняется в составе его молекулы, обогащенной щелочью плавня, и в раствор не переходит. Содержание двуокиси кремния определяют колориметрическим методом по реакции с молибденовой синью.
Чувствительность определения — 5 мкг двуокиси кремния в анализируемом объеме раствора.
Силикаты не мешают определению.
Аппаратура см. выше.
Реактивы
1. Стандартный раствор двуокиси кремния. Навеску кварца х.ч. в количестве 0,05 г тщательно растирают (до состояния пудры) в агатовой ступке, перемешивают с 2,5 г составного плавня из солей калия или с 2 г плавня из солей натрия и сплавляют в серебряном или платиновом тигле. Сплавление проводят в муфельной печи при температуре 800 — 850 °C. Однородная масса в тигле расплавляется до жидкого состояния и выдерживается при этой температуре в течение 2 — 3 мин. Затем после охлаждения остывшую массу в том же тигле обрабатывают постепенно 40 мл 5% раствора карбоната натрия при кипении. Каждую порцию раствора из тигля переводят по палочке в воронку с плотным фильтром, в мерную колбу емкостью 100 мл. Фильтрат доводят водой до 100 мл. В исходном растворе содержится 0,5 мг/мл двуокиси кремния. Рабочий раствор с содержанием 100 мкг/мл готовят путем разбавления водой исходного раствора. Стандартные растворы должны быть свежеприготовленными.
2. Бикарбонат калия или натрия, х.ч., безводный.
3. Хлорид калия или натрия х.ч., безводный.
4. Составной плавень. Равные весовые количества тонкорастертых бикарбоната и хлорида калия (или бикарбоната и хлорида натрия) тщательно перемешивают и смесь растирают в агатовой ступке небольшими порциями, которые затем соединяют вместе и снова растирают и перемешивают. Готовый составной плавень сохраняют в банке с пришлифованной пробкой.
Однородность плавня проверяют 3 раза титрованием по 1 г плавня, растворенного в 10 мл воды 1 н. раствором соляной кислоты. На 1 г плавня должно быть израсходовано 5 мл соляной кислоты. Для сплавления применяют 50-кратное количество (к навеске пробы) плавня из солей калия или 40-кратное количество плавня из солей натрия.
5. Соляная кислота, 1 н. раствор.
6. Карбонат натрия кристаллический, 5 и 10% растворы, свежеприготовленные.
7. Соляная кислота, уд. вес 1,19, разбавленная 1:3.
8. Соляная кислота, уд. вес 1,19, разбавленная 1:1.
9. Азотная кислота, концентрированная, разбавленная 1:1.
10. Хлорид аммония, 2% раствор.
11. Уксусная кислота, 10% раствор.
12. Молибдат аммония [(NH4)6Mo7O24·4H2O], 10% раствор, готовят из перекристаллизованной соли. Для перекристаллизации готовят насыщенный раствор молибдата аммония в воде, фильтруют, выливают в равный объем этилового спирта, добавляют несколько капель аммиака до явного запаха, фильтруют на воронке Бюхнера и сушат на воздухе.
13. Виннокаменная кислота, 10% раствор.
14. Сульфат натрия кристаллический, насыщенный раствор на холоду, свежеприготовленный.
Отбор пробы
Для определения свободной двуокиси кремния можно использовать пробы пыли, отобранные на бумажный фильтр или фильтр АФА-ХТ, помещенный в патрон. Скорость отбора до 100 л/мин. (для определения двуокиси кремния навеска должна быть не менее 20 мг).
Ход анализа
Фильтр с пылью взвешивают и сжигают в муфельной печи при температуре 600 °C, остаток взвешивают.
Если в пыли отсутствуют несиликатные соединения металлов (Fe, Ca, Mg и др.) — окиси, гидроокиси или карбонаты, пробу, растертую до состояния пудры в агатовой ступке, сплавляют непосредственно с плавнем. Если указанные вещества присутствуют, пробу подвергают предварительной обработке кислотами. Для этого пробу помещают в стакан и обрабатывают 10 мл смеси из равных объемов соляной и азотной кислот, разведенных 1:1, при кипении в течение 2 мин. Охлажденный раствор фильтруют и фильтр с осадком обрабатывают дважды по 10 мл нагретым до кипения 2% раствором хлорида аммония, затем 20 мл кипящего 10% раствора карбоната натрия и снова дважды по 10 мл кипящего 2% раствора хлорида аммония.
Затем фильтр с осадком подсушивают и помещают в серебряный или платиновый тигель, сжигают и прокаливают в муфельной печи. Сплавление пробы с плавнем производят следующим образом: берут навеску составного плавня 2,5 г из солей калия и 2 г из солей натрия. Из этой навески вносят небольшое количество плавня в охлажденный тигель с пробой, перемешивают палочкой и переносят в агатовую ступку. Тигель дважды обрабатывают оставшимся плавнем, который переносят в агатовую ступку и все растирают до состояния пудры. Полученную смесь снова вносят в тигель, остатком плавня обрабатывают агатовую ступку и также переносят в тигель. Содержимое тигля тщательно перемешивают и тигель помещают в муфельную печь для сплавления, постепенно нагревая печь до 800 — 900 °C. После полного расплавления массы тигель выдерживают при этой температуре еще в течение 2 — 5 мин. Для извлечения кремниевой кислоты застывший плав обрабатывают 40 мл 5% раствора карбоната натрия при нагревании; для этого небольшими порциями вносят раствор карбоната натрия в тигель, нагревают почти до кипения и фильтруют через плотный фильтр. Тигель промывают водой, которую фильтруют через тот же фильтр, и доводят объем фильтра до 100 мл дистиллированной водой. Если раствор мутный, то его фильтруют еще раз через тот же фильтр.
Для анализа в колориметрические пробирки вносят 1 и 5 мл пробы. Готовят шкалу стандартов (табл. 108).
Таблица 108
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
|
Рабочий стандартный раствор, мл |
0 |
0,05 |
0,1 |
0,2 |
0,3 |
0,4 |
0,6 |
0,8 |
1 |
|
5% раствор карбоната натрия, мл |
5 |
4,95 |
4,9 |
4,8 |
4,7 |
4,6 |
4,4 |
4,2 |
4 |
|
Содержание двуокиси кремния, мкг |
0 |
5 |
10 |
20 |
30 |
40 |
60 |
80 |
100 |
Во все пробирки проб и шкалы добавляют по 0,5 мл соляной кислоты (1:3) и 0,5 мл 10% раствора уксусной кислоты, взбалтывают, приливают по 1 мл 10% раствора молибдата аммония, снова взбалтывают и оставляют на 15 мин. По истечении этого времени добавляют по 0,5 мл 10% раствора виннокаменной кислоты, взбалтывают и оставляют на 5 мин., затем приливают по 1 мл насыщенного раствора сульфита натрия, взбалтывают и ставят на 5 мин. на водяную баню, нагретую до 70 °C. Через 5 мин. пробирки охлаждают и сравнивают интенсивность окраски проб со шкалой стандартов.
Расчет
Количество двуокиси кремния (X) в процентах вычисляют по формуле:
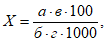
где а — количество двуокиси кремния, найденное в анализируемом объеме пробы (мкг); б — объем раствора, взятый для анализа (мл); в — общий объем пробы (мл); г — навеска пыли (мг); 1/1000 — коэффициент пересчета микрограммов в миллиграммы; 100 — коэффициент пересчета найденного количества двуокиси кремния в проценты.
ЛИТЕРАТУРА
1. Гельденскиольд Р.С., Минаев А.А. Гравиметрический метод определения пыли в атмосферном воздухе с использованием фильтра из ткани ФПП-15-1,5. Гиг. и сан., 1968, 1.
2. Определение двуокиси кремния и силикатов. В кн.: Е.А. Перегуд, Е.В. Гернет. Химический анализ воздуха промышленных предприятий. Л., «Химия», 1973.
3. Панин К.П. Определение сажи в атмосферном воздухе. Гиг. и сан., 1968, 12.
4. Полежаев Н.Г. Определение свободной двуокиси кремния в присутствии силикатов в промышленных выбросах и атмосферной пыли. Гиг. и сан., 1957, 11.
5. Рязанов В.А. Определение сажи в воздухе. В сб.: Труды Пермского медицинского института, 1934, т. VII.