Чтобы разобраться, что такое генетическая мутация, вспомним, как устроены ДНК и гены.
ДНК (дезоксирибонуклеиновая кислота) — это длинная молекула, которую принято называть «двойной спиралью». Она хранит биологическую информацию, которая «записана» в виде генетического кода.
Ген — это основная «единица» наследственной информации. Он представляет собой кусочек ДНК.

Главный редактор, заведующий хирургическим отделением
Задать вопрос
Врач-хирург высшей квалификационной категории, доктор медицинских наук, профессор кафедры общей хирургии АГМУ.
Содержание
- Какова функция генов?
- Что такое мутация?
- Мутации: хорошие, плохие, нейтральные
- «Хорошие» мутации
- «Плохие» мутации
- «Нейтральные» мутации
- Что делать, если генетический тест показал мутацию?
- Мутации при раке
- Как часто в клетках тела человека происходят мутации?
- Почему мутации приводят к онкологическим заболеваниям?
- Протоонкогены
- Гены-супрессоры опухолевого роста
- Гены репарации ДНК
- Что способствует развитию мутаций, которые приводят к раку?
- Распространенные мутации при раке
- Почему важно изучать мутации при онкологических заболеваниях?
- Как определяют мутации при раке?
- Что такое эпигенетические изменения, и какую роль они играют в онкологии?
- Новости «Евроонко»
- Родословная нейронов: как носить в себе множество мутаций и выглядеть совершенно здоровым
- Автор
- Редактор
- Мутации: патология или норма?
- Сколько мутаций может содержать в себе геном нейрона?
- «Генеалогическое древо» нейронов
- «Движение — это смерть»
- Наследственные болезни человека
- Наследственные болезни человека. Классификация.
- Хромосомные болезни
- Генные болезни
- Заболевания с наследственной предрасположенностью или полигенные болезни
- Диагностика наследственных болезней
- Лечение наследственных болезней
- Читайте также:
- Как влияют соматические мутации на здоровье людей
- Что такое мозаицизм? Соматический мозаицизм и мозаицизм по половым клеткам
- Соматический мозаицизм
- Мозаицизм по половым клеткам
Какова функция генов?
В части генов в виде кода записаны «рецепты» изготовления белков. Именно белки выполняют основные функции для поддержания жизнедеятельности организма: они отвечают за пищеварение, кровообращение, иммунитет, передачу информации между клетками.
Код представляет собой последовательность нуклеотидов.
В нашей ДНК есть четыре азотистых основания:
Основания одной цепи соединяются с основаниями другой цепи парами (аденин с тимином, цитозин с гуанином).

Какие способности передаются по наследству?
Если посмотреть на двойную спираль ДНК, то ее горизонтальные «ступени» будут парами оснований, а вертикальные боковые части — сахарами и фосфатами.
Чтобы изготовить белки по записанному в генах коду, специальные соединения — ферменты — «читают» и копируют код. В результате получаются длинные одноцепочечные молекулы — РНК (рибонуклеиновые кислоты), но это еще не белок. РНК лишь несут в себе информацию о первичной структуре белка, поэтому их называют матричными (сокращенно — мРНК). Эти молекулы покидают ядро клетки и перемещаются в ее цитоплазму. Там специальные органы — рибосомы — считывают код мРНК и изготавливают по этому «рецепту» белок.
Что такое мутация?
Генетическая мутация — это любое изменение в нуклеотидной последовательности ДНК.
К основным типам мутаций относятся:
- транзиция — замена аденина на гуанин или замена тимина на цитозин;
- трансверсия — аденин или гуанин меняются местами с тимином или цитозином;
- делеция — потеря участка ДНК;
- инсерция — добавление участка ДНК;
- дупликация — удвоение участка ДНК;
- инверсия — изменение, при котором участок хромосомы поворачивается на 180°;
- транслокация — мутация, при которой хромосомы обмениваются фрагментами.
Мутации могут происходить по разным причинам.
Спонтанные генетические мутации
Они происходят на протяжении всей нашей жизни. Можно сказать, что это нормальное явление, которое случается в ходе разных процессов, например, при копировании ДНК.
Как правило, такие ошибки не грозят серьезными последствиями, потому что у нашего организма есть механизмы защиты.

Как связаны спортивные достижения и генетика
К ним относится, например, апоптоз — процесс программируемой гибели «испорченной» клетки, или репарация — починка нити ДНК. В этом случае ошибочный участок ДНК вырезается, а на его месте формируется новый.
Мутации, вызванные внешним влиянием
Мутации могут возникнуть под воздействием внешних неблагоприятных факторов, например, химических веществ, ионизирующего излучения или заражения вирусами.
Белки, которые отвечают за исправление ошибок, как правило, могут исправить испорченные цепи ДНК или привести одну хромосому в соответствие с другой. Но, если ошибки произошли на уровне генома или количества хромосом, защитные механизмы будут бессильны.
Наследственные генетические мутации
Такие мутации достаются человеку от родителей. Бывают случаи, когда генетическое нарушение передается из поколения в поколение (как, например, болезнь гемофилия), иногда мутации происходят в яйцеклетках и сперматозоидах и таким образом передаются ребенку.
Бывают случаи, когда мутации возникают на этапе формирования зиготы — клетки, которая образуется в результате оплодотворения. Как и в предыдущем случае, механизмы репарации с такими мутациями работают далеко не всегда, а ряд заболеваний и вовсе связан с нарушениями в процессе починки (например, пигментная ксеродерма — заболевание кожи, представляющее собой повышенную чувствительность к ультрафиолету).
Мутации: хорошие, плохие, нейтральные
Не все генетические мутации опасны. Важно понимать, что именно мутации объясняют генетические различия между видами. Изменения генов влекут за собой изменение характеристик организма, и в результате этого он может стать либо более, либо менее приспособленным к выживанию.
В ходе естественного отбора преимущество получают те живые существа, которые обладают более «полезным» набором характеристик, и тогда мутация закрепляется в популяции, становясь нормой.
«Хорошие» мутации
Ученым известно, что, например, у людей с определенным вариантом гена GPR75 риск ожирения снижен на 54%. А те, у кого есть хотя бы одна копия такого варианта гена, имеют более низкий индекс массы тела.

Мутации генов могут давать человеку и другие преимущества: так, мутировавший ген EPOR дал финскому лыжнику, трехкратному олимпийскому чемпиону Ээро Мянтюранта высокую чувствительность к эритропоэтину — гормону, который помогает нашим клеткам поддерживать оптимальный уровень кислорода и выводить углекислый газ. Это изменило и объем красных кровяных клеток в крови спортсмена, и объем кислорода, который эти клетки способны переносить. В результате Мянтюранта получил супервыносливость — его организм легко справлялся с повышенной потребностью в кислороде во время физических нагрузок.
«Плохие» мутации
Генетические мутации могут вызывать различные заболевания. Например, изменения гена DMD вызывают дистрофию Дюшенна — нервно-мышечное заболевание, которое проявляется у мужчин намного чаще, чем у женщин. А к серповидноклеточной анемии — нарушению в строении белка гемоглобина, который переносит кислород от легких к органам, — приводят мутации гена HBB. Хорея Гентингтона — тяжелое заболевание нервной системы — развивается из-за мутации в гене HTT.
Однако далеко не всегда генетическое заболевание связано с мутацией одного гена. Так, синдром Дауна возникает из-за изменения количества хромосом — в клетках пациентов с этой болезнью 47 хромосом вместо обычных 46.
Ряд заболеваний, таких как рак, диабет, расстройства аутического спектра, появляются из-за комбинации факторов. Пациенты могут иметь генетическую предрасположенность, но значительную роль играют и внешние факторы — неправильный образ жизни, неблагоприятная окружающая среда.
«Нейтральные» мутации
Нейтральные мутации, как следует из названия, не оказывают на здоровье человека ни положительного, ни отрицательного эффекта. Как правило, эти мутации затрагивают нуклеотидную последовательность ДНК, но не сказываются на строении белков.
Так происходит, потому что наш генетический код обладает так называемой избыточностью — это значит, что ряд аминокислот кодируется несколькими способами, чтобы случайные ошибки при копировании с меньшей вероятностью привели к нарушению функции или отсутствию кодируемого белка.
Бывает и так, что мутация гена все-таки меняет аминокислоту. Тем не менее, это не всегда приводит к нарушению функции белка.
Что делать, если генетический тест показал мутацию?
Во многих случаях наличие генетических нарушений не означает, что у человека непременно разовьется какое-то заболевание. Это значит лишь то, что пациент обладает генетическим вариантом, который чаще встречается у людей с этим заболеванием, и, вероятно, предрасположен к возникновению проблемы больше остальных.
Важнейшую роль в таких случаях играют внешние факторы: образ жизни, привычки, окружающая среда.
Ученые сходятся на том, что важными условиями сохранения здоровья являются:
● сбалансированное питание, богатое овощами и фруктами;
● регулярные занятия спортом;
● отказ от курения и алкоголя;
● достаточное количество сна.
Эти правила помогут значительно снизить риск развития таких распространенных заболеваний, как рак, проблемы с сердечно-сосудистой системой, диабет второго типа.

Генная терапия: шанс или фантастика?
В том случае, если у человека есть мутация, связанная с моногенным заболеванием (то есть таким, которое возникает из-за «поломки» всего лишь одного гена), то существует риск, что он передаст этот вариант гена своему ребенку. Кроме того, болезнь может проявиться и у самого обладателя «плохого» гена — в этом случае ему следует обратиться к специалистам. Как правило, генетические заболевания не лечатся, но врач сможет порекомендовать препараты или изменения образа жизни (например, диета), чтобы уменьшить проявления болезни.
Результаты Генетического теста Атлас подскажут персональные рекомендации по улучшению образа жизни, которые помогут минимизировать риск появления заболеваний. Используя эти знания, будет проще спланировать подходящий рацион, спортивные нагрузки и тренировки, профилактические обследования.
Мутации при раке
Тело человека состоит примерно из 37 триллионов клеток. Информация о строении и функциях каждой из них закодирована в ДНК. Любая злокачественная опухоль является результатом нарушения работы тех или иных генов, а главная причина этого кроется в мутациях. Некоторые из них человек получает с рождения, и они присутствуют во всех клетках тела. А некоторые возникают уже в течение жизни под влиянием тех или иных факторов — эти мутации будут обнаруживаться только в потомках той клетки, в которой изначально возникла «поломка».
На этой странице мы собрали всю информацию о генетических нарушениях, связанных с онкологическими заболеваниями, представленную на нашем сайте.

Как часто в клетках тела человека происходят мутации?
Мутагенез — процесс непрерывный. Он происходит на всех этапах развития любого организма: в половых клетках, с самых первых дней существования эмбриона и на протяжении всей жизни. К счастью, далеко не все мутации вредны. Многие из них нейтральные (то есть не приносят ни вреда, ни пользы), а некоторые даже дают организму определенные преимущества.
Мутации — это главный двигатель эволюции живых организмов. В 2018 году были опубликованы результаты исследования, во время которого ученые обнаружили, что у 20-летних людей на одну клетку слизистой оболочки пищевода в среднем приходится по 100 мутаций, а у людей более старшего возраста — по 2000. Большинство из них не опасны, но некоторые затрагивают онкогены.
Чаще всего рак связан именно с соматическими, приобретенными, мутациями. Согласно современным представлениям, наследственные мутации ответственны за развитие лишь 5–10% онкопатологий. А по результатам исследования, опубликованного в 2020 году, наследственные мутации, связанные с раком, встречаются у каждого восьмого онкологического больного.
Почему мутации приводят к онкологическим заболеваниям?
Конечно же, далеко не все мутации и далеко не во всех генах приводят к развитию онкологических заболеваний. Чтобы нормальная клетка стала злокачественной, нарушения должны произойти в определенных генах:
Протоонкогены
Это гены, которые в результате мутаций способны превращаться в онкогены. В свою очередь, онкогены — это дефектные гены, которые способствуют развитию злокачественной опухоли, например, путем бесконтрольного размножения клеток. Характерный пример — EGFR.
Гены-супрессоры опухолевого роста
В норме они «сдерживают» клетки и не дают им стать злокачественными. Когда в этих генах возникают мутации, они перестают выполнять свои функции. Например, к этой категории относится ген TP53, кодирующий белок p53.
Гены репарации ДНК
Чаще всего их относят к генам-супрессорам опухолевого роста, но иногда выделяют в отдельную группу. Белки, кодируемые этими генами, исправляют «ошибки», возникающие в ДНК. Например, продукты генов BRCA1 и BRCA2 восстанавливают двухцепочечные разрывы в ДНК путем гомологичной рекомбинации — процесса, при котором поврежденная хромосома использует свою «сестру-близнеца» в качестве шаблона для репарации. Когда эти гены перестают правильно работать из-за мутаций, ДНК не может нормально восстанавливаться, и в ней накапливается еще больше повреждений.

Что способствует развитию мутаций, которые приводят к раку?
Мутации, связанные с онкозаболеваниями, бывают двух основных видов. Наследственные мутации происходят в половых клетках, и затем они будут присутствовать во всех клетках тела ребенка. Соматические мутации присутствуют только в клетках, в которых они изначально возникли, и в их потомках — например, только в злокачественной опухоли.
Обычно, чтобы нормальная клетка превратилась в злокачественную, в ней должен возникнуть целый набор мутаций. В каждом конкретном случае невозможно точно сказать, что именно послужило причиной. Скорее всего, единой причины и нет. На организм человека постоянно действует множество факторов, и многие из них могут способствовать поломкам в генах.
Вот список некоторых распространенных факторов риска, способствующих развитию рака:
Некоторые инфекции, например, ВПЧ
Неблагоприятная экологическая ситуация, воздействие вредных веществ на работе
Пол — многие онкологические заболевания чаще встречаются у мужчин или женщин
Семейный анамнез: рак у близких родственников
Большое количество красного и обработанного мяса (говядина, свинина, баранина, фастфуд, сосиски и колбасы, бекон и пр.)
Распространенные мутации при раке
Мутации в гене EGFR — белка-рецептора эпидермального фактора роста, который находится на поверхности клеток и активирует их размножение
T790M — один из вариантов мутации в гене EGFR
Мутации в гене ROS1 — белка, который встроен в клеточную мембрану и передает сигналы, играющие роль в росте и дифференцировке клеток
Мутации в гене BRAF. Белок, который он кодирует, участвует в регуляции делений клеток путем активации специфического сигнального пути.
Слияние генов с участием NTRK — когда из двух генов получается “неправильный”, гибридный. Гены NTRK кодируют белки Trk, которые выполняют разные функции, в том числе защищают клетки от апоптоза.
Мутации в гене ALK — белка, встроенного в клеточную мембрану, который передает сигналы, связанные с ростом, миграцией клеток, образованием новых кровеносных сосудов
Мутации в генах BRCA — белков, которые помогают восстанавливать ДНК, когда в обеих ее цепочках происходят разрывы
Мутации в генах RAS — белков, которые передают сигналы внутри клеток и регулируют клеточные деления. Семейство RAS включает три гена: KRAS, NRAS и HRAS.
Мутации в PIK3CA — гене, который кодирует белок PI3K, участвующий в регуляции важных процессов в клетках
Мутации в HRR — группе генов, продукты которых участвуют в репарации ДНК при двухцепочечных разрывах
Мутации в TP53 — гене, кодирующем белок p53, «страж генома», который останавливает размножение клеток с поврежденной ДНК и «приказывает» им совершить «самоубийство».
Результатом некоторых мутаций может стать микросателлитная нестабильность — состояние, при котором нарушается восстановление ДНК, и она приобретает повышенную склонность к мутациям.
Почему важно изучать мутации при онкологических заболеваниях?
Для врачей-онкологов важно знать, какие мутации произошли в раковых клетках у конкретного пациента. Это помогает решать важные задачи:
- судить о степени агрессивности рака, выстраивать прогноз;
- определять тип, подтип некоторых злокачественных опухолей;
- подбирать наиболее эффективные противоопухолевые препараты;
- назначать персонализированную терапию при запущенном раке, когда не помогают стандартные схемы лечения из протоколов.
Выявление мутаций, связанных с раком, у здоровых людей помогает оценивать риск развития онкологического заболевания, проводить профилактику и решать, кому назначать дополнительные скрининговые исследования.
А ученым знания о мутациях в опухолевых клетках помогают создавать новые лекарства.


Как определяют мутации при раке?
В федеральной сети клиник экспертной онкологии «Евроонко» доступны все современные исследования для выявления мутаций при раке:
Что такое эпигенетические изменения, и какую роль они играют в онкологии?
Не меньшую (а может быть, даже и более важную) роль, чем мутации, в развитии рака играют эпигенетические изменения. Этим термином называют такие модификации, которые не меняют последовательность генетического кода, но влияют на активность генов.
Чаще всего встречаются две разновидности эпигенетических изменений (но есть и другие):
- Метилирование ДНК — присоединение к ее определенным участкам метильных групп. Чаще всего они заставляют «молчать» определенные гены. В норме у человека метилирован 1% всего генома. В некоторых раковых клетках этот показатель ниже. За счет этого в них могут «включаться» онкогены.
- Модификации гистонов. ДНК организована таким образом, что напоминает бусы — эта структура называется нуклеосомой. В качестве бусинок выступают особые белки — гистоны. Они обмотаны нитями ДНК и влияют на активность генов. Даже небольшие изменения в гистонах могут сильно повлиять на регуляцию работы генов, заставить некоторые из них «замолчать» или, напротив, активировать.

Эпигенетика — очень интересная наука. Возможно, со временем она поможет ученым создать еще больше эффективных препаратов для лечения рака.
Новости «Евроонко»

Почему у многих курильщиков не развивается рак легких? 20 апреля 2022

С возрастом у людей накапливается много мутаций, способных привес. 27 декабря 2021

Как родинка превращается в меланому? 08 декабря 2021
Лечение пациентов проводится в соответствии со стандартами и рекомендациями наиболее авторитетных онкологических сообществ. «Евроонко» является партнёром Фонда борьбы с раком. ВНИМАНИЮ ПАЦИЕНТОВ: Рекомендации по лечению даются только после консультации у специалиста. Ваши персональные данные обрабатываются на сайте в целях его корректного функционирования. Если вы не согласны с обработкой ваших персональных данных, просим вас покинуть сайт. Оставаясь на сайте, вы даёте согласие на обработку ваших персональных данных.
Политика конфиденциальности © ООО «Центр инновационных медицинских технологий». 2012 — 2022
Товарный знак зарегистрирован. Все права защищены. Незаконное использование преследуется по закону.




Содержание данного интернет ресурса (сайт https://www.euroonco.ru/), включая любую информацию и результаты интеллектуальной деятельности, защищены законодательством Российской Федерации и международными соглашениями. Любое использование, копирование, воспроизведение или распространение любой размещенной информации, материалов и (или) их частей не допускается без предварительного получения согласия правообладателя и влечет применение мер ответственности.
Сведения и материалы, размещенные на сайте , подготовлены исключительно в информационных целях и не являются медицинской консультацией или заключением. Авторы информационных материалов сайта не могут гарантировать применимость такой информации для целей третьих лиц и не несут ответственности за решения третьих лиц и связанные с ними возможные прямые или косвенные потери и/или ущерб, возникшие в результате использования информации или какой-либо ее части, содержащейся на сайте.
Сайт использует файлы cookies для правильного функционирования, индивидуального подбора контента в социальных сетях и сбора анонимной статистики о пользователях с помощью систем аналитики для повышения удобства использования. Оставаясь на сайте, вы соглашаетесь с правилами использования файлов cookies.
Родословная нейронов: как носить в себе множество мутаций и выглядеть совершенно здоровым

Автор
Редактор
Статья на конкурс «био/мол/текст»: На протяжении долгой истории развития нейробиологии ученые придерживались догмы: мозг взрослого человека не подвержен изменениям. Однако в ходе нового исследования впервые было показано, что значительное количество мутаций присутствует в мозговом веществе абсолютно здоровых людей, причем чаще всего они обнаруживаются в генах, которые нейрон использует наиболее активно. Попробуем разобраться, как этим можно воспользоваться и чем это грозит.
Обратите внимание!
Эта работа опубликована в номинации «лучшее новостное сообщение» конкурса «био/мол/текст»-2015.
Спонсором номинации «Лучшая статья о механизмах старения и долголетия» является фонд «Наука за продление жизни». Спонсором приза зрительских симпатий выступила фирма Helicon.
Спонсоры конкурса: Лаборатория биотехнологических исследований 3D Bioprinting Solutions и Студия научной графики, анимации и моделирования Visual Science.
Мутации: патология или норма?
Каждая клетка нашего тела была создана путем деления клеток-предшественниц, которые, в свою очередь, восходят в развитии к зиготе. Значит ли это, что общий путь развития всех клеток организма обеспечивает общность генетического материала? Нет, и виной тому — мутации (рис. 1).

Рисунок 1. «Древо развития» мутаций в организме человека. Нарушения, обнаруженные в коре головного мозга, часто встречаются и в периферических органах. Рисунок из [4].
Мутации — коварные преобразования ДНК, которые страшны тем, что могут возникать в клетках любых тканей многоклеточного организма и на любых стадиях его развития. Распространено мнение, что мутации опасны потому, что могут наследоваться потомством. Действительно, мутации, передающиеся по наследству, приводят к возникновению и развитию таких серьезных заболеваний нервной системы, как шизофрения, аутизм, болезнь Альцгеймера. Виной тому — приобретаемые детьми генетические нарушения половых клеток родителей. Однако существуют и другие, ненаследуемые мутации, которые возникают в соматических клетках человека на протяжении всей его жизни.
Большинство людей имеет определенное количество соматических мутаций. Известным примером следствий соматических мутаций является появление опухолевых клеток, для которых характерны генетические нарушения*. Однако далеко не всегда соматическая мутация приводит к развитию онкологических заболеваний. Часто изменения генома не выливаются в какие-либо серьезные заболевания и могут встречаться у полностью здоровых людей. До настоящего момента ученые точно не знали, накапливаются ли они в головном мозге в таком количестве, чтобы послужить причиной серьезных нарушений нервной системы.
По мере роста и взросления человека геномы нейронов его головного мозга накапливают существенные различия. К такому выводу пришли ученые Бостонской детской больницы (Boston Children’s Hospital) и Гарвардской медицинской школы (Harvard Medical School), опровергнув утверждение, что мозг взрослого человека не изменяется в течение жизни* [4, 5].
* — Последние годы оказались особенно урожайными на опровержения железобетонных нейробиологических догм. Как нам на радость разобрались с приговором «нервные клетки не восстанавливаются», описано в статье «Всё, что вы всегда хотели знать о взрослом нейрогенезе, но боялись спросить» [6]. — Ред.
Результаты недавнего исследования показали, что значительное количество соматических мутаций можно обнаружить в мозге полностью здоровых людей. Так, со временем геномы нейронов головного мозга человека начинают различаться — появляется мозаицизм. Это научное открытие позволит изучать роль соматических мутаций отдельных нейронов в развитии человека и ряда нервно-психических заболеваний.
Сколько мутаций может содержать в себе геном нейрона?
Ранее не было точно известно, способны ли соматические мутации, возникающие в нейронах головного мозга, провоцировать возникновение и развитие нейродегенеративных заболеваний. Для того чтобы установить истину, ученые решили изучить особую разновидность мутаций — однонуклеотидные варианты (single-nucleotide variants, SNVs). Эти нарушения могут возникнуть в нескольких или даже всего в одной клетке головного мозга. Исследователи проанализировали 36 нейронов, взятых из головного мозга трех умерших людей: 15-летней девушки, 17-летнего юноши и 42-летней женщины, которые не страдали нейродегенеративными заболеваниями.
Используя методы капиллярной цифровой полимеразной цепной реакции (digital PCR) и секвенирования геномов единичных клеток [7], ученые обнаружили, что каждый отдельный нейрон из трех образцов ткани мозга содержит в среднем от 1468 до 1580 однонуклеотидных вариантов (рис. 2). И если появление SNVs в опухолевых клетках связано преимущественно с ошибками при репликации ДНК, то нейронные мутации возникают в основном вследствие активной транскрипции генов.

Рисунок 2. Карта мутаций генома корковых нейронов одного человека. 136 нейронов головного мозга 17-летнего человека распределены по четырем группам (обозначены разными цветами), выделенным по одной или нескольким мутациям (буквами A-D обозначены 18 клональных соматических мутаций). Рисунок из [5].
Дополнительно ученые сравнили гены нервных клеток с генетическим материалом, взятым из других тканей — в частности, сердца и кожи. Этот анализ показал, что мутации в нейронах в целом совпадают с однонуклеотидными вариантами в других типах клеток, то есть такие мутации присутствуют и в нейронах, и в других частях организма человека. Более того, был установлен следующий интересный факт: в ряде случаев клетки коры мозга показывали более высокую степень родства не с соседними нейронами, а с другими клетками организма (например, кардиомиоцитами).
Также было проведено исследование нервных клеток, взятых из разных областей головного мозга, с целью обнаружения аналогичных мутаций. Полученные результаты позволили сделать предположение о происхождении нервных клеток.
«Генеалогическое древо» нейронов

Рисунок 3. «Родословное древо» человека из книги Э. Геккеля «Антропогения». Идея объединения всех живых существ в единое «древо» имеет более чем 150-летнюю историю. Рисунок с сайта vivovoco.astronet.ru.
Мутации возникают как за счет ошибок копирования ДНК, которые потенциально могут сопровождать каждый репликационный цикл, так и в результате иных мутационных процессов — например, под действием ультрафиолетового света. Закономерное следствие — каждая клетка организма может иметь свой собственный уникальный геном, который несет в себе информацию о происхождении и развитии клетки, воздействии на нее внешних факторов. Такие «записи» онтогенеза отдельных клеток позволят создать их «родословное древо».
В разных клетках происходят разные мутации, что обеспечивает несходство геномов. Кроме этого, мутационный профиль несет в себе долговременную память о происхождении и развитии каждой клетки. Информация, полученная при секвенировании геномов индивидуальных нейронов, может быть использована для декодирования всей картины развития человеческого мозга — для реконструкции своеобразного «генеалогического древа» нейронов. Этот подход позволит расширить знания о природе возрастных заболеваний и выявить различия между мозгом человека и мозгом других животных.
Основоположником генеалогии можно считать Чарльза Дарвина, который впервые изобразил филогенетическое древо живых организмов еще в 1837 году. В его основу легла идея о том, что все виды живых существ связаны друг с другом общим происхождением, подобно ветвям дерева, которые объединяет общий корень (рис. 3). Подобные мысли использовали при создании клеточной теории ученые Т. Шванн и М. Шлейден, определившие клетку как единый структурный элемент всех живых организмов. Наконец, более чем через 150 лет, в 2005 году, Д. Фрумкин и соавторы в своем исследовании показали, что соматические мутации присутствуют в клетках в достаточном количестве и могут быть использованы для воссоздания взаимосвязей всех клеток человека [8]. Таким образом, далеко не свежие идеи лежат в основе нового заключения о том, что каждый человек несет в себе собственное (клеточное) генеалогическое древо*.
* — Дерево — это красиво и понятно, дерево — это аллегория из мира эукариот. А как же работают биологи с прокариотическими дебрями, где схемы родственных связей не то что дерево не напоминают, даже лес для них простоват — сеть да и только? Об эволюционных перипетиях в разных мирах читайте: «Эволюция между молотом и наковальней, или как микробиология спасла эволюцию от поглощения молекулярной биологией» [9], «Карл Вёзе (1928–2012)» [10], «Вирусные геномы в системе эволюции» [11] и «Закинули археи эволюционный невод и вытянули. » [12]. — Ред.
Кристофер Уолш и другие сотрудники Гарвардской медицинской школы в результате исследования однонуклеотидных вариантов предложили подход к установлению происхождения нервных клеток человека [5]. Так, если в двух отдельно взятых нейронах присутствуют одни и те же мутации, то они с высокой долей вероятности происходят от одной клетки-предшественницы. В том случае, если совпадает лишь часть мутаций, пути развития нейронов в какой-то момент времени разошлись.
Сравнивая геномы нейронов и других клеток организма, можно сделать следующий вывод: если какая-то мутация присутствует и в головном мозге, и в других соматических клетках — она возникла на раннем этапе онтогенеза. Если же определенная мутация встречается лишь в некоторых нейронах, это говорит о том, что она появилась сравнительно недавно. Таким образом можно проследить «родословную» нейронов вплоть до конкретного дня эмбрионального развития.
«Движение — это смерть»
Выше упоминалось, что мутации, обеспечивающие различия геномов соматических клеток, могут быть вызваны многими факторами. Так, длительное время считалось, что основной причиной мутаций в клетках головного мозга являются ошибки репликации ДНК. Однако в результате настоящего исследования ученые установили, что нарушения возникают не во время деления клетки, а при экспрессии генов. Всем известный девиз «Движение — это жизнь» не работает в случае соматических мутаций нейронов. Исследователи установили, что каждый раз, когда гены нейронов нашего мозга начинают активно работать — запуская программу синтеза новых белков, — появляется определенный риск возникновения мутаций.
Ученые пришли к выводу о том, что мутации в головном мозге накапливаются с возрастом и могут быть причастны к развитию нейродегенеративных заболеваний. Получается, что любой человек, сколь бы здоровым он ни был, является носителем огромного количества соматических мутаций — своеобразных «факторов риска». Чем это реально может грозить и как этого избежать — покажет время и будущие исследования нейробиологов.
Наследственные болезни человека
Наследственные болезни человека это заболевания, связанные с нарушением работы наследственного аппарата клеток и передающиеся по наследству от родителей потомству. Основной резервуар генетической информации находится в ядерных хромосомах. Все клетки человеческого организма содержат в ядрах одинаковое количество хромосом. Исключение составляют половые клетки или гаметы — сперматозоиды и яйцеклетки, и малая часть клеток, которые делятся прямым делением. Меньшая доля генетической информации содержится в митохондриальной ДНК.
Наследственные болезни человека. Классификация.
Патология генетического аппарата бывает на хромосомном уровне, на уровне отдельного гена, а также бывает связана с дефектом или отсутствием нескольких генов. Наследственные болезни человека подразделяются на:
Хромосомные болезни
Наиболее известны хромосомные заболевания по типу трисомии — дополнительной третьей хромосомы в паре:
- Синдром Дауна — трисомия по 21 паре;
- Синдром Патау — трисомия по 13 паре;
- Синдром Эдвардса — трисомия по 18 паре хромосом.

Синдром Шерешевского — Тёрнера обусловлен отсутствием одной Х-хромосомы у женщин.
Синдром Кляйнфельтера — дополнительная Х-хромосома у мужчин.
Другие хромосомные болезни связаны со структурной перестройкой хромосом при их нормальном количестве. Например, потеря или удвоение части хромосомы, обмен участками хромосом из разных пар.

Патогенез хромосомных болезней не совсем ясен. По-видимому, срабатывает механизм «пятого колеса», когда отсутствие или лишняя хромосома в паре мешает нормальной работе генетического аппарата в клетках.
Генные болезни
Причины наследственных заболеваний на генном уровне заключаются в повреждении части ДНК, в результате которого возникает дефект одного определенного гена. Чаще всего генные мутации ответственны за наследственные дегенеративные заболевания или наследственные болезни обмена веществ в результате нарушения синтеза соответствующего структурного белка или белка-фермента:
- Муковисцидоз;
- Гемофилия;
- Фенилкетонурия;
- Альбинизм; ;
- Серповидноклеточная анемия;
- Непереносимость лактозы;
- Другие обменные заболевания.
Моногенные наследственные заболевания наследуются по классическим законам Грегора Менделя. Различают аутосомно-доминантный, аутосомно-рецессивный и сцепленный с полом типы наследования.

При близкородственных браках чаще всего реализуется именно генный тип наследственных заболеваний.
Заболевания с наследственной предрасположенностью или полигенные болезни
К ним относятся:
- ; ;
- Ишемическая болезнь сердца;
- Ревматоидный полиартрит;
- Рак молочной железы;
- Псориаз;
- Шизофрения;
- Аллергические заболевания;
- Язвенная болезнь желудка…
Список можно продолжать и дальше. Найдется лишь малая часть болезней, которые так или иначе не связаны с наследственной предрасположенностью. Действительно, все процессы функционирования организма обусловлены синтезом разнообразных белков, как строительных, так и белков-ферментов.

Но если при моногенных наследственных болезней за синтез соответствующего белка отвечает один ген, то при полигенных наследственных заболеваниях за сложный метаболический процесс отвечают несколько разных генов. Поэтому мутация одного из них может быть компенсированной и проявляться только при дополнительных внешних неблагоприятных условиях. Этим объясняется, что у больных данными заболеваниями дети болеют ими не всегда, и, наоборот, у здоровых родителей дети могут болеть этими болезнями. Поэтому в случае полигенных наследственных заболеваний можно говорить лишь о большей или меньшей предрасположенности.
Диагностика наследственных болезней
Методы диагностики наследственных болезней:
- . Большинство хромосомных и генных заболеваний диагностируются по внешним или клиническим признакам. Характерный внешний вид при синдроме Дауна, полидактилия при синдроме Патау, отсутствие пигментации при альбинизме, тяжелые формы дыхательной недостаточности при муковисцидозе.
- Генеалогический метод заключается в построении генеалогического древа на основании данных анамнеза. Позволяет рассчитать вероятность развития генных заболеваний у детей при болезни или носительстве мутировавших генов у родителей и предков.
- Лабораторная и инструментальная диагностика. Наследственные болезни человека, связанные на нарушением метаболизма, выявляются с помощью клинических анализов. Например, серповидноклеточная анемия по общему анализу крови, определением фенилаланина при фенилкетоурии, нарушение коагулограммы при гемофилии. При мраморной болезни выявляются характерные рентгенологические изменения костей, при гемофилии — гемартрозы.
- Цитогенетическое исследование идентифицирует количество и строение хромосом. Применяется для диагностики хромосомных болезней.
- Скрининг на наследственные заболевания ориентирован на выявление генетической патологии на доклиническом уровне. Это комплексный метод, заключающийся в проведении просеивающего теста на некоторые наследственные заболевания: муковисцидоз, фенилкетонурия, болезнь Тея-Сакса и некоторых других редких наследственных заболеваний.
- Пренатальная диагностика наследственных заболеваний — метод выявления наследственной патологии на стадии внутриутробного развития.
- Молекулярно-цитогенетические и молекулярно-биологические методы позволяют провести диагностику наследственных болезней на уровне дефекта гена. Перспективное направление, однако, оно значительно осложняется при полигенных наследственных заболеваниях, когда за проявление болезни отвечают множество разных генов. Даже при моногенных заболеваниях не всегда известен и идентифицирован ответственный ген, что также затрудняет диагностику.
- Методы генетического выявления предрасположенности и профилактика наследственных заболеваний в онкологии. В 2006 году в США была основана частная компания «23andMe». Главное направление деятельности компании — выявление степени предрасположенности к некоторым заболеваниям, в частности к раку молочной железы и яичников на основе анализа генов BRCA1 и BRCA2. В значительной мере интерес к этой теме был подогрет в 2013 году операцией по удалению груди известной голливудской актрисе А. Джоли.

Однако, следует учитывать, что мутации генов BRCA1 и BRCA2 ответственны за рак молочной железы (РМЖ) только в 5-10%, а их наличие или отсутствие лишь изменяет степень риска достаточно редкой формы РМЖ. Расчет эффективности этого метода будет представлен в следующих публикациях.
Лечение наследственных болезней
Симптоматическое лечение заключается в коррекции метаболических и других патологических нарушений, связанных с данным заболеванием.
Диетотерапия направлена на исключение продуктов, содержащих вещества, которые не усваиваются или не переносятся больными.
Генотерапия направлена на введение в генетический аппарат клеток человека, эмбриона или зиготы генетического материала, компенсирующего дефекты мутированных генов. Успехи генотерапии пока невелики. Но медицина с оптимизмом смотрит на развитие генноинженерных методов в терапии наследственных заболеваний.
Читайте также:
Антибиотики в продуктах
В мире от электрического тока ежегодно погибает 50 тысяч, а всего в ХХ веке от электричества погибло 2 или 3 миллиона человек. Интересно, придет ли кому-то в голову запр.
Отрицательный резус-фактор у женщин
Легкое конспирологическое чтиво о связи группы крови или резус-фактора с убывающим Сатурном и судьбой нисколько не отменяют проблему резус-конфликта и гемолитическую желт.
Дальтонизм или цветовая слепота
Ночью все кошки серы! Действительно, при недостатке освещения цветовые рецепторы сетчатки глаза не работают, и мы видим лишь оттенки серого. Но стоит взойти солнцу, и кра.
Как влияют соматические мутации на здоровье людей
Что такое мозаицизм? Соматический мозаицизм и мозаицизм по половым клеткам
Мозаицизм — присутствие в организме или ткани по крайней мере двух генетически отличающихся клеточных линий, производных от одной зиготы. Хотя мы имеем обыкновение считать, что при формировании клеток они получают одинаковый набор генов и хромосом, это упрощенное представление. Мы уже ввели понятие мозаицизма, вызванного инактивацией Х-хромосомы, формирующей две различных популяции соматических клеток у женщин, с активной отцовской или материнский Х-хромосомой.
Чаще мутации, возникающие в единственной клетке во внутриутробной или послеродовой жизни, могут вызывать линии клеток, генетически отличающихся от зиготы, поскольку однажды произошедшая мутация может передаваться всем потомкам клетки. Мозаицизм по числовым или структурным аномалиям хромосом — клинически важный феномен, а соматические мутации признают основными причинами многих типов опухолей.
Мозаицизм по мутациям в одном гене, в соматических или половых клетках, объясняет множество необычных клинических наблюдений, например сегментный нейрофиброматоз, когда кожные проявления появляются не по всему телу, а участками, или повторное рождение у здоровых родителей двух или более детей с несовершенным остеогенезом, высокопенетрантной аутосомно-доминантной болезнью.
Популяция клеток, несущих мутацию у мозаичного пациента, теоретически может присутствовать в некоторых тканях тела, но не в гаметах (чистый соматический мозаицизм), ограничиваться только гаметами (чистый половой мозаицизм) или присутствовать как в соматических, так и в половых клетках, в зависимости от того, когда произошла мутация в ходе эмбрионального развития. Включает ли мозаицизм только соматические ткани, только половые клетки или и те, и другие, зависит от времени появления мутации в эмбриогенезе — до или после разделения половых и соматических клеток.
Если до, то и соматические, и половые клетки будут мозаичными, а мутация может передаваться потомству и проявляться соматически в мозаичной форме. Мутацию, произошедшую позже, обнаруживают только в половых клетках или части соматических тканей. Таким образом, например, если мутация произошла в предшественнике половых клеток, часть гамет будет нести мутацию. До мейоза половые клетки проходят около 30 митотических делений у женщин и несколько сотен у мужчин, допуская массу возможностей для мутаций, происходящих в течение митотических этапов развития гаметы.
Выявление мозаицизма по мутации только в половых или соматических клетках может быть трудным, поскольку отсутствие мутации в клетках из легкодоступных соматических тканей (например, лейкоцитов периферической крови, кожи или клеток слизистой оболочки рта) не доказывает, что мутация не присутствует где-нибудь еще, включая половые клетки. Охарактеризовать распространенность соматического мозаицизма еще труднее, если мутантный аллель у мозаичного эмбриона встречается исключительно во внезародышевых тканях (т.е. в плаценте) и не присутствует в самом эмбрионе.

Соматический мозаицизм
Мутации, влияющие на морфогенез и проявляющиеся в ходе эмбрионального развития, могут быть обнаружены как сегментные или пятнистые аномалии, в зависимости от этапа, в котором произошла мутация, и происхождения соматической клетки. Например, нейрофиброматоз I типа иногда может проявляться как сегментный, влияя только на одну часть тела. Сегментный нейрофиброматоз I типа вызван мозаицизмом по мутации, произошедшей после зачатия. В таких случаях родители пациента здоровы, но если он (или она) рожает больного ребенка, фенотип у ребенка полный, т.е. не сегментный.
В таких случаях мутация находится в гаметах пациента и, по-видимому, произошла до разделения половой и соматической линии клеток.
Мозаицизм по половым клеткам
Так как шанс, что аутосомное или Х-сцепленное заболевание, вызванное новой мутацией, может неоднократно происходить в сибстве, очень низок, поскольку спонтанные мутации обычно происходят редко (порядка 1 на 104-106), появление двух независимых мутаций в том же гене в одной семье весьма маловероятно (менее чем 1 на 108-1012). После тщательного исключения даже малых проявлений болезни у здоровых родителей ребенка с аутосомно-доминантным или Х-сцепленным заболеванием и при отрицательных результатах молекулярного тестирования носительства обычно принято сообщать родителям, что болезнь их ребенка — результат новой мутации и шанс того же дефекта у последующего ребенка незначительный, равный популяционному риску.
Существуют, тем не менее, хорошо подтвержденные примеры, когда фенотипически здоровые родители с отрицательными тестами на носительство имеют более чем одного ребенка с высокопенетрантным аутосомно-доминантным или Х-сцепленным заболеванием. Такие необычные родословные могут объясняться половым мозаицизмом. Половой мозаицизм хорошо подтвержден почти в 6% летальных форм аутосомно-доминантного несовершенного остеогенеза, когда мутации в гене коллагена I типа приводят к формированию аномального коллагена, ломким костям и частым переломам.
Родословные, которые могут объясняться половым мозаицизмом, также отмечены при нескольких других заболеваниях, например гемофилии А, гемофилии В и мышечной дистрофии Дюшенна, но очень редко встречаются при других доминантных болезнях, например ахондроплазии. Точно измерить частоту полового мозаицизма сложно, но приблизительно считают, что самая высокая встречаемость отмечена при мышечной дистрофии Дюшенна, при которой до 15% матерей в изолированных случаях не имеют подтверждения мутации в их соматических тканях при наличии мутации в половых клетках.
Теперь, когда феномен полового мозаицизма признан, генетики и генетические консультанты отдают себе отчет о потенциальной погрешности прогноза, что специфический аутосомно-доминантный или Х-сцепленный фенотип, кажущийся новой мутацией, имеет незначительный риск повторения в потомстве. Очевидно, для болезней с доказанной возможностью полового мозаицизма фенотипически здоровым родителям ребенка, у которых предположительно болезнь возникла вследствие новой мутации, нужно сообщать, что риск повторения не настолько незначительный.
Кроме того, родители ребенка с любым аутосомно-доминантным или Х-сцепленным заболеванием имеют риск повторения 3-4%, даже если половой мозаицизм не доказан и если известно, что они не носители мутации. Таким парам следует предложить доступную пренатальную диагностику. Точный риск повторения оценить трудно, поскольку он зависит от доли мутантных гамет.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021

A red tulip exhibiting a partially yellow petal due to a mutation in its genes

Mutation with double bloom in the Langheck Nature Reserve near Nittel, Germany
In biology, a mutation is an alteration in the nucleic acid sequence of the genome of an organism, virus, or extrachromosomal DNA.[1] Viral genomes contain either DNA or RNA. Mutations result from errors during DNA or viral replication, mitosis, or meiosis or other types of damage to DNA (such as pyrimidine dimers caused by exposure to ultraviolet radiation), which then may undergo error-prone repair (especially microhomology-mediated end joining),[2] cause an error during other forms of repair,[3][4] or cause an error during replication (translesion synthesis). Mutations may also result from insertion or deletion of segments of DNA due to mobile genetic elements.[5][6][7]
Mutations may or may not produce detectable changes in the observable characteristics (phenotype) of an organism. Mutations play a part in both normal and abnormal biological processes including: evolution, cancer, and the development of the immune system, including junctional diversity. Mutation is the ultimate source of all genetic variation, providing the raw material on which evolutionary forces such as natural selection can act.
Mutation can result in many different types of change in sequences. Mutations in genes can have no effect, alter the product of a gene, or prevent the gene from functioning properly or completely. Mutations can also occur in non-genic regions. A 2007 study on genetic variations between different species of Drosophila suggested that, if a mutation changes a protein produced by a gene, the result is likely to be harmful, with an estimated 70% of amino acid polymorphisms that have damaging effects, and the remainder being either neutral or marginally beneficial.[8] Due to the damaging effects that mutations can have on genes, organisms have mechanisms such as DNA repair to prevent or correct mutations by reverting the mutated sequence back to its original state.[5]
Overview[edit]
Mutations can involve the duplication of large sections of DNA, usually through genetic recombination.[9] These duplications are a major source of raw material for evolving new genes, with tens to hundreds of genes duplicated in animal genomes every million years.[10] Most genes belong to larger gene families of shared ancestry, detectable by their sequence homology.[11] Novel genes are produced by several methods, commonly through the duplication and mutation of an ancestral gene, or by recombining parts of different genes to form new combinations with new functions.[12][13]
Here, protein domains act as modules, each with a particular and independent function, that can be mixed together to produce genes encoding new proteins with novel properties.[14] For example, the human eye uses four genes to make structures that sense light: three for cone cell or color vision and one for rod cell or night vision; all four arose from a single ancestral gene.[15] Another advantage of duplicating a gene (or even an entire genome) is that this increases engineering redundancy; this allows one gene in the pair to acquire a new function while the other copy performs the original function.[16][17] Other types of mutation occasionally create new genes from previously noncoding DNA.[18][19]
Changes in chromosome number may involve even larger mutations, where segments of the DNA within chromosomes break and then rearrange. For example, in the Homininae, two chromosomes fused to produce human chromosome 2; this fusion did not occur in the lineage of the other apes, and they retain these separate chromosomes.[20] In evolution, the most important role of such chromosomal rearrangements may be to accelerate the divergence of a population into new species by making populations less likely to interbreed, thereby preserving genetic differences between these populations.[21]
Sequences of DNA that can move about the genome, such as transposons, make up a major fraction of the genetic material of plants and animals, and may have been important in the evolution of genomes.[22] For example, more than a million copies of the Alu sequence are present in the human genome, and these sequences have now been recruited to perform functions such as regulating gene expression.[23] Another effect of these mobile DNA sequences is that when they move within a genome, they can mutate or delete existing genes and thereby produce genetic diversity.[6]
Nonlethal mutations accumulate within the gene pool and increase the amount of genetic variation.[24] The abundance of some genetic changes within the gene pool can be reduced by natural selection, while other «more favorable» mutations may accumulate and result in adaptive changes.

For example, a butterfly may produce offspring with new mutations. The majority of these mutations will have no effect; but one might change the color of one of the butterfly’s offspring, making it harder (or easier) for predators to see. If this color change is advantageous, the chances of this butterfly’s surviving and producing its own offspring are a little better, and over time the number of butterflies with this mutation may form a larger percentage of the population.
Neutral mutations are defined as mutations whose effects do not influence the fitness of an individual. These can increase in frequency over time due to genetic drift. It is believed that the overwhelming majority of mutations have no significant effect on an organism’s fitness.[25][26] Also, DNA repair mechanisms are able to mend most changes before they become permanent mutations, and many organisms have mechanisms for eliminating otherwise-permanently mutated somatic cells.
Beneficial mutations can improve reproductive success.[27][28]
Causes[edit]
Four classes of mutations are (1) spontaneous mutations (molecular decay), (2) mutations due to error-prone replication bypass of naturally occurring DNA damage (also called error-prone translesion synthesis), (3) errors introduced during DNA repair, and (4) induced mutations caused by mutagens. Scientists may also deliberately introduce mutant sequences through DNA manipulation for the sake of scientific experimentation.
One 2017 study claimed that 66% of cancer-causing mutations are random, 29% are due to the environment (the studied population spanned 69 countries), and 5% are inherited.[29]
Humans on average pass 60 new mutations to their children but fathers pass more mutations depending on their age with every year adding two new mutations to a child.[30]
Spontaneous mutation[edit]
Spontaneous mutations occur with non-zero probability even given a healthy, uncontaminated cell. Naturally occurring oxidative DNA damage is estimated to occur 10,000 times per cell per day in humans and 100,000 times per cell per day in rats.[31] Spontaneous mutations can be characterized by the specific change:[32]
- Tautomerism – A base is changed by the repositioning of a hydrogen atom, altering the hydrogen bonding pattern of that base, resulting in incorrect base pairing during replication.[33] Theoretical results suggest that proton tunneling is an important factor in the spontaneous creation of GC tautomers.[34]
- Depurination – Loss of a purine base (A or G) to form an apurinic site (AP site).
- Deamination – Hydrolysis changes a normal base to an atypical base containing a keto group in place of the original amine group. Examples include C → U and A → HX (hypoxanthine), which can be corrected by DNA repair mechanisms; and 5MeC (5-methylcytosine) → T, which is less likely to be detected as a mutation because thymine is a normal DNA base.
- Slipped strand mispairing – Denaturation of the new strand from the template during replication, followed by renaturation in a different spot («slipping»). This can lead to insertions or deletions.
Error-prone replication bypass[edit]
There is increasing evidence that the majority of spontaneously arising mutations are due to error-prone replication (translesion synthesis) past DNA damage in the template strand. In mice, the majority of mutations are caused by translesion synthesis.[35] Likewise, in yeast, Kunz et al.[36] found that more than 60% of the spontaneous single base pair substitutions and deletions were caused by translesion synthesis.
Errors introduced during DNA repair[edit]
Although naturally occurring double-strand breaks occur at a relatively low frequency in DNA, their repair often causes mutation. Non-homologous end joining (NHEJ) is a major pathway for repairing double-strand breaks. NHEJ involves removal of a few nucleotides to allow somewhat inaccurate alignment of the two ends for rejoining followed by addition of nucleotides to fill in gaps. As a consequence, NHEJ often introduces mutations.[37]

Induced mutation[edit]
Induced mutations are alterations in the gene after it has come in contact with mutagens and environmental causes.
Induced mutations on the molecular level can be caused by:
- Chemicals
- Hydroxylamine
- Base analogs (e.g., Bromodeoxyuridine (BrdU))
- Alkylating agents (e.g., N-ethyl-N-nitrosourea (ENU). These agents can mutate both replicating and non-replicating DNA. In contrast, a base analog can mutate the DNA only when the analog is incorporated in replicating the DNA. Each of these classes of chemical mutagens has certain effects that then lead to transitions, transversions, or deletions.
- Agents that form DNA adducts (e.g., ochratoxin A)[39]
- DNA intercalating agents (e.g., ethidium bromide)
- DNA crosslinkers
- Oxidative damage
- Nitrous acid converts amine groups on A and C to diazo groups, altering their hydrogen bonding patterns, which leads to incorrect base pairing during replication.
- Radiation
- Ultraviolet light (UV) (including non-ionizing radiation). Two nucleotide bases in DNA—cytosine and thymine—are most vulnerable to radiation that can change their properties. UV light can induce adjacent pyrimidine bases in a DNA strand to become covalently joined as a pyrimidine dimer. UV radiation, in particular longer-wave UVA, can also cause oxidative damage to DNA.[40]
- Ionizing radiation. Exposure to ionizing radiation, such as gamma radiation, can result in mutation, possibly resulting in cancer or death.
Whereas in former times mutations were assumed to occur by chance, or induced by mutagens, molecular mechanisms of mutation have been discovered in bacteria and across the tree of life. As S. Rosenberg states, «These mechanisms reveal a picture of highly regulated mutagenesis, up-regulated temporally by stress responses and activated when cells/organisms are maladapted to their environments—when stressed—potentially accelerating adaptation.»[41] Since they are self-induced mutagenic mechanisms that increase the adaptation rate of organisms, they have some times been named as adaptive mutagenesis mechanisms, and include the SOS response in bacteria,[42] ectopic intrachromosomal recombination[43] and other chromosomal events such as duplications.[41]
Classification of types[edit]
By effect on structure[edit]
![]()
Five types of chromosomal mutations
![]()
Types of small-scale mutations
The sequence of a gene can be altered in a number of ways.[44] Gene mutations have varying effects on health depending on where they occur and whether they alter the function of essential proteins.
Mutations in the structure of genes can be classified into several types.
Large-scale mutations[edit]
Large-scale mutations in chromosomal structure include:
- Amplifications (or gene duplications) or repetition of a chromosomal segment or presence of extra piece of a chromosome broken piece of a chromosome may become attached to a homologous or non-homologous chromosome so that some of the genes are present in more than two doses leading to multiple copies of all chromosomal regions, increasing the dosage of the genes located within them.
- Polyploidy, duplication of entire sets of chromosomes, potentially resulting in a separate breeding population and speciation.
- Deletions of large chromosomal regions, leading to loss of the genes within those regions.
- Mutations whose effect is to juxtapose previously separate pieces of DNA, potentially bringing together separate genes to form functionally distinct fusion genes (e.g., bcr-abl).
- Large scale changes to the structure of chromosomes called chromosomal rearrangement that can lead to a decrease of fitness but also to speciation in isolated, inbred populations. These include:
- Chromosomal translocations: interchange of genetic parts from nonhomologous chromosomes.
- Chromosomal inversions: reversing the orientation of a chromosomal segment.
- Non-homologous chromosomal crossover.
- Interstitial deletions: an intra-chromosomal deletion that removes a segment of DNA from a single chromosome, thereby apposing previously distant genes. For example, cells isolated from a human astrocytoma, a type of brain tumor, were found to have a chromosomal deletion removing sequences between the Fused in Glioblastoma (FIG) gene and the receptor tyrosine kinase (ROS), producing a fusion protein (FIG-ROS). The abnormal FIG-ROS fusion protein has constitutively active kinase activity that causes oncogenic transformation (a transformation from normal cells to cancer cells).
- Loss of heterozygosity: loss of one allele, either by a deletion or a genetic recombination event, in an organism that previously had two different alleles.
Small-scale mutations[edit]
Small-scale mutations affect a gene in one or a few nucleotides. (If only a single nucleotide is affected, they are called point mutations.) Small-scale mutations include:
- Insertions add one or more extra nucleotides into the DNA. They are usually caused by transposable elements, or errors during replication of repeating elements. Insertions in the coding region of a gene may alter splicing of the mRNA (splice site mutation), or cause a shift in the reading frame (frameshift), both of which can significantly alter the gene product. Insertions can be reversed by excision of the transposable element.
- Deletions remove one or more nucleotides from the DNA. Like insertions, these mutations can alter the reading frame of the gene. In general, they are irreversible: Though exactly the same sequence might, in theory, be restored by an insertion, transposable elements able to revert a very short deletion (say 1–2 bases) in any location either are highly unlikely to exist or do not exist at all.
- Substitution mutations, often caused by chemicals or malfunction of DNA replication, exchange a single nucleotide for another.[45] These changes are classified as transitions or transversions.[46] Most common is the transition that exchanges a purine for a purine (A ↔ G) or a pyrimidine for a pyrimidine, (C ↔ T). A transition can be caused by nitrous acid, base mispairing, or mutagenic base analogs such as BrdU. Less common is a transversion, which exchanges a purine for a pyrimidine or a pyrimidine for a purine (C/T ↔ A/G). An example of a transversion is the conversion of adenine (A) into a cytosine (C). Point mutations are modifications of single base pairs of DNA or other small base pairs within a gene. A point mutation can be reversed by another point mutation, in which the nucleotide is changed back to its original state (true reversion) or by second-site reversion (a complementary mutation elsewhere that results in regained gene functionality). As discussed below, point mutations that occur within the protein coding region of a gene may be classified as synonymous or nonsynonymous substitutions, the latter of which in turn can be divided into missense or nonsense mutations.
By impact on protein sequence[edit]

![]()
Point mutations classified by impact on protein
![]()
The effect of a mutation on protein sequence depends in part on where in the genome it occurs, especially whether it is in a coding or non-coding region. Mutations in the non-coding regulatory sequences of a gene, such as promoters, enhancers, and silencers, can alter levels of gene expression, but are less likely to alter the protein sequence. Mutations within introns and in regions with no known biological function (e.g. pseudogenes, retrotransposons) are generally neutral, having no effect on phenotype – though intron mutations could alter the protein product if they affect mRNA splicing.
Mutations that occur in coding regions of the genome are more likely to alter the protein product, and can be categorized by their effect on amino acid sequence:
- A frameshift mutation is caused by insertion or deletion of a number of nucleotides that is not evenly divisible by three from a DNA sequence. Due to the triplet nature of gene expression by codons, the insertion or deletion can disrupt the reading frame, or the grouping of the codons, resulting in a completely different translation from the original.[48] The earlier in the sequence the deletion or insertion occurs, the more altered the protein produced is. (For example, the code CCU GAC UAC CUA codes for the amino acids proline, aspartic acid, tyrosine, and leucine. If the U in CCU was deleted, the resulting sequence would be CCG ACU ACC UAx, which would instead code for proline, threonine, threonine, and part of another amino acid or perhaps a stop codon (where the x stands for the following nucleotide).) By contrast, any insertion or deletion that is evenly divisible by three is termed an in-frame mutation.
- A point substitution mutation results in a change in a single nucleotide and can be either synonymous or nonsynonymous.
- A synonymous substitution replaces a codon with another codon that codes for the same amino acid, so that the produced amino acid sequence is not modified. Synonymous mutations occur due to the degenerate nature of the genetic code. If this mutation does not result in any phenotypic effects, then it is called silent, but not all synonymous substitutions are silent. (There can also be silent mutations in nucleotides outside of the coding regions, such as the introns, because the exact nucleotide sequence is not as crucial as it is in the coding regions, but these are not considered synonymous substitutions.)
- A nonsynonymous substitution replaces a codon with another codon that codes for a different amino acid, so that the produced amino acid sequence is modified. Nonsynonymous substitutions can be classified as nonsense or missense mutations:
- A missense mutation changes a nucleotide to cause substitution of a different amino acid. This in turn can render the resulting protein nonfunctional. Such mutations are responsible for diseases such as Epidermolysis bullosa, sickle-cell disease, and SOD1-mediated ALS.[49] On the other hand, if a missense mutation occurs in an amino acid codon that results in the use of a different, but chemically similar, amino acid, then sometimes little or no change is rendered in the protein. For example, a change from AAA to AGA will encode arginine, a chemically similar molecule to the intended lysine. In this latter case the mutation will have little or no effect on phenotype and therefore be neutral.
- A nonsense mutation is a point mutation in a sequence of DNA that results in a premature stop codon, or a nonsense codon in the transcribed mRNA, and possibly a truncated, and often nonfunctional protein product. This sort of mutation has been linked to different diseases, such as congenital adrenal hyperplasia. (See Stop codon.)
By effect on function[edit]
A mutation becomes an effect on function mutation when the exactitude of functions between a mutated protein and its direct interactor undergoes change. The interactors can be other proteins, molecules, nucleic acids, etc. There are many mutations that fall under the category of by effect on function, but depending on the specificity of the change the mutations listed below will occur.[50]
- Loss-of-function mutations, also called inactivating mutations, result in the gene product having less or no function (being partially or wholly inactivated). When the allele has a complete loss of function (null allele), it is often called an amorph or amorphic mutation in Muller’s morphs schema. Phenotypes associated with such mutations are most often recessive. Exceptions are when the organism is haploid, or when the reduced dosage of a normal gene product is not enough for a normal phenotype (this is called haploinsufficiency). A disease that is caused by a loss-of-function mutation is Gitelman syndrome and cystic fibrosis.[51]
- Gain-of-function mutations also called activating mutations, change the gene product such that its effect gets stronger (enhanced activation) or even is superseded by a different and abnormal function. When the new allele is created, a heterozygote containing the newly created allele as well as the original will express the new allele; genetically this defines the mutations as dominant phenotypes. Several of Muller’s morphs correspond to the gain of function, including hypermorph (increased gene expression) and neomorph (novel function). In December 2017, the U.S. government lifted a temporary ban implemented in 2014 that banned federal funding for any new «gain-of-function» experiments that enhance pathogens «such as Avian influenza, SARS, and the Middle East Respiratory Syndrome or MERS viruses. Many diseases are caused by this mutation including systemic mastocytosis and STAT3 disease.[52]
- Dominant negative mutations (also called anti-morphic mutations) have an altered gene product that acts antagonistically to the wild-type allele. These mutations usually result in an altered molecular function (often inactive) and are characterized by a dominant or semi-dominant phenotype. In humans, dominant negative mutations have been implicated in cancer (e.g., mutations in genes p53, ATM, CEBPA, and PPARgamma]). Marfan syndrome is caused by mutations in the FBN1 gene, located on chromosome 15, which encodes fibrillin-1, a glycoprotein component of the extracellular matrix. Marfan syndrome is also an example of dominant negative mutation and haploinsufficiency.
- Lethal mutations result in the instant death of the developing organism. Lethal mutations can also lead to a substantial loss in the life expectancy of the organism. An example of a disease that is caused by a dominant lethal mutation is Huntington’s disease.
- Null mutations, also known as Amorphic mutations, are a form of loss-of-function mutations that completely prohibit the gene’s function. The mutation leads to a complete loss of operation at the phenotypic level, also causing no gene product to be formed. Atopic eczema and dermatitis syndrome are common diseases caused by a null mutation of the gene that activates filaggrin.
- Suppressor mutations are a type of mutation that causes the double mutation to appear normally. In suppressor mutations the phenotypic activity of a different mutation is completely suppressed, thus causing the double mutation to look normal. There are two types of suppressor mutations, there are intragenic and extragenic suppressor mutations. Intragenic mutations occur in the gene where the first mutation occurs, while extragenic mutations occur in the gene that interacts with the product of the first mutation. A common disease that results from this type of mutation is Alzheimer’s disease.[53]
- Neomorphic mutations are a part of the gain-of-function mutations and are characterized by the control of new protein product synthesis. The newly synthesized gene normally contains a novel gene expression or molecular function. The result of the neomorphic mutation is the gene where the mutation occurs has a complete change in function.[54]
- A back mutation or reversion is a point mutation that restores the original sequence and hence the original phenotype.[55]
By effect on fitness (harmful, beneficial, neutral mutations)[edit]
In genetics, it is sometimes useful to classify mutations as either harmful or beneficial (or neutral):
- A harmful, or deleterious, mutation decreases the fitness of the organism. Many, but not all mutations in essential genes are harmful (if a mutation does not change the amino acid sequence in an essential protein, it is harmless in most cases).
- A beneficial, or advantageous mutation increases the fitness of the organism. Examples are mutations that lead to antibiotic resistance in bacteria (which are beneficial for bacteria but usually not for humans).
- A neutral mutation has no harmful or beneficial effect on the organism. Such mutations occur at a steady rate, forming the basis for the molecular clock. In the neutral theory of molecular evolution, neutral mutations provide genetic drift as the basis for most variation at the molecular level. In animals or plants, most mutations are neutral, given that the vast majority of their genomes is either non-coding or consists of repetitive sequences that have no obvious function («junk DNA»).[56]
Large-scale quantitative mutagenesis screens, in which thousands of millions of mutations are tested, invariably find that a larger fraction of mutations has harmful effects but always returns a number of beneficial mutations as well. For instance, in a screen of all gene deletions in E. coli, 80% of mutations were negative, but 20% were positive, even though many had a very small effect on growth (depending on condition).[57] Note that gene deletions involve removal of whole genes, so that point mutations almost always have a much smaller effect. In a similar screen in Streptococcus pneumoniae, but this time with transposon insertions, 76% of insertion mutants were classified as neutral, 16% had a significantly reduced fitness, but 6% were advantageous.[58]
This classification is obviously relative and somewhat artificial: a harmful mutation can quickly turn into a beneficial mutations when conditions change. Also, there is a gradient from harmful/beneficial to neutral, as many mutations may have small and mostly neglectable effects but under certain conditions will become relevant. Also, many traits are determined by hundreds of genes (or loci), so that each locus has only a minor effect. For instance, human height is determined by hundreds of genetic variants («mutations») but each of them has a very minor effect on height,[59] apart from the impact of nutrition. Height (or size) itself may be more or less beneficial as the huge range of sizes in animal or plant groups shows.
Distribution of fitness effects (DFE)[edit]
Attempts have been made to infer the distribution of fitness effects (DFE) using mutagenesis experiments and theoretical models applied to molecular sequence data. DFE, as used to determine the relative abundance of different types of mutations (i.e., strongly deleterious, nearly neutral or advantageous), is relevant to many evolutionary questions, such as the maintenance of genetic variation,[60] the rate of genomic decay,[61] the maintenance of outcrossing sexual reproduction as opposed to inbreeding[62] and the evolution of sex and genetic recombination.[63] DFE can also be tracked by tracking the skewness of the distribution of mutations with putatively severe effects as compared to the distribution of mutations with putatively mild or absent effect.[64] In summary, the DFE plays an important role in predicting evolutionary dynamics.[65][66] A variety of approaches have been used to study the DFE, including theoretical, experimental and analytical methods.
- Mutagenesis experiment: The direct method to investigate the DFE is to induce mutations and then measure the mutational fitness effects, which has already been done in viruses, bacteria, yeast, and Drosophila. For example, most studies of the DFE in viruses used site-directed mutagenesis to create point mutations and measure relative fitness of each mutant.[67][68][69][70] In Escherichia coli, one study used transposon mutagenesis to directly measure the fitness of a random insertion of a derivative of Tn10.[71] In yeast, a combined mutagenesis and deep sequencing approach has been developed to generate high-quality systematic mutant libraries and measure fitness in high throughput.[72] However, given that many mutations have effects too small to be detected[73] and that mutagenesis experiments can detect only mutations of moderately large effect; DNA sequence analysis can provide valuable information about these mutations.
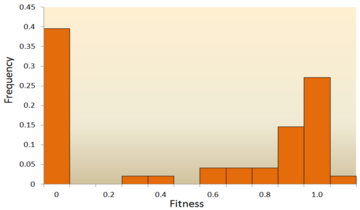
The distribution of fitness effects (DFE) of mutations in vesicular stomatitis virus. In this experiment, random mutations were introduced into the virus by site-directed mutagenesis, and the fitness of each mutant was compared with the ancestral type. A fitness of zero, less than one, one, more than one, respectively, indicates that mutations are lethal, deleterious, neutral, and advantageous.[67]
-

This figure shows a simplified version of loss-of-function, switch-of-function, gain-of-function, and conservation-of-function mutations.
Molecular sequence analysis: With rapid development of DNA sequencing technology, an enormous amount of DNA sequence data is available and even more is forthcoming in the future. Various methods have been developed to infer the DFE from DNA sequence data.[74][75][76][77] By examining DNA sequence differences within and between species, we are able to infer various characteristics of the DFE for neutral, deleterious and advantageous mutations.[24] To be specific, the DNA sequence analysis approach allows us to estimate the effects of mutations with very small effects, which are hardly detectable through mutagenesis experiments.

One of the earliest theoretical studies of the distribution of fitness effects was done by Motoo Kimura, an influential theoretical population geneticist. His neutral theory of molecular evolution proposes that most novel mutations will be highly deleterious, with a small fraction being neutral.[25][78] A later proposal by Hiroshi Akashi proposed a bimodal model for the DFE, with modes centered around highly deleterious and neutral mutations.[79] Both theories agree that the vast majority of novel mutations are neutral or deleterious and that advantageous mutations are rare, which has been supported by experimental results. One example is a study done on the DFE of random mutations in vesicular stomatitis virus.[67] Out of all mutations, 39.6% were lethal, 31.2% were non-lethal deleterious, and 27.1% were neutral. Another example comes from a high throughput mutagenesis experiment with yeast.[72] In this experiment it was shown that the overall DFE is bimodal, with a cluster of neutral mutations, and a broad distribution of deleterious mutations.
Though relatively few mutations are advantageous, those that are play an important role in evolutionary changes.[80] Like neutral mutations, weakly selected advantageous mutations can be lost due to random genetic drift, but strongly selected advantageous mutations are more likely to be fixed. Knowing the DFE of advantageous mutations may lead to increased ability to predict the evolutionary dynamics. Theoretical work on the DFE for advantageous mutations has been done by John H. Gillespie[81] and H. Allen Orr.[82] They proposed that the distribution for advantageous mutations should be exponential under a wide range of conditions, which, in general, has been supported by experimental studies, at least for strongly selected advantageous mutations.[83][84][85]
In general, it is accepted that the majority of mutations are neutral or deleterious, with advantageous mutations being rare; however, the proportion of types of mutations varies between species. This indicates two important points: first, the proportion of effectively neutral mutations is likely to vary between species, resulting from dependence on effective population size; second, the average effect of deleterious mutations varies dramatically between species.[24] In addition, the DFE also differs between coding regions and noncoding regions, with the DFE of noncoding DNA containing more weakly selected mutations.[24]
By inheritance[edit]

A mutation has caused this moss rose plant to produce flowers of different colors. This is a somatic mutation that may also be passed on in the germline.
In multicellular organisms with dedicated reproductive cells, mutations can be subdivided into germline mutations, which can be passed on to descendants through their reproductive cells, and somatic mutations (also called acquired mutations),[86] which involve cells outside the dedicated reproductive group and which are not usually transmitted to descendants.
Diploid organisms (e.g., humans) contain two copies of each gene—a paternal and a maternal allele. Based on the occurrence of mutation on each chromosome, we may classify mutations into three types. A wild type or homozygous non-mutated organism is one in which neither allele is mutated.
- A heterozygous mutation is a mutation of only one allele.
- A homozygous mutation is an identical mutation of both the paternal and maternal alleles.
- Compound heterozygous mutations or a genetic compound consists of two different mutations in the paternal and maternal alleles.[87]
Germline mutation[edit]
A germline mutation in the reproductive cells of an individual gives rise to a constitutional mutation in the offspring, that is, a mutation that is present in every cell. A constitutional mutation can also occur very soon after fertilisation, or continue from a previous constitutional mutation in a parent.[88] A germline mutation can be passed down through subsequent generations of organisms.
The distinction between germline and somatic mutations is important in animals that have a dedicated germline to produce reproductive cells. However, it is of little value in understanding the effects of mutations in plants, which lack a dedicated germline. The distinction is also blurred in those animals that reproduce asexually through mechanisms such as budding, because the cells that give rise to the daughter organisms also give rise to that organism’s germline.
A new germline mutation not inherited from either parent is called a de novo mutation.
Somatic mutation[edit]
GENE MUTATIONS:
Gene mutations include either the replacement of one of the nucleotides with the nucleotide by the other nucleotide or may be by the addition or the deletion of the nucleotide.[89] This would be explained as the sudden change or the alteration in nucleotide sequence of the DNA molecule, which would affect one pair of nucleotide or the bigger art of the gene on chromosome.[90] These gene mutations can be further classified as:
1. Point mutations: This results when there is difference in only one base pair of nucleotide which can also be called as base pair substitution and this is also one of the common type among the gene mutations. Point mutations can be again divided into three types of mutations namely Silent mutations, Nonsense mutations, Missense mutations.
a) Silent Mutations:
This occurs when there is a change in codon for one amino acid molecule is swapped or is into the other codon of the same amino acid molecule and is also referred as “synonymous mutations”
b) Missense Mutations:
This occurs when the codon of one amino acid is interchanged with the codon of another amino acid and can also be referred as non-synonymous mutations.
c) Nonsense Mutations:
This occurs when the codon of the amino acid changes to the stop codon.
2. Frameshift Mutations:
This kind of mutation results when there is addition or deletion of DNA base molecules changes the reading frame of the gene. This mutations would be insertions or deletions.[90]
a) Insertion:
This type of mutation differs the DNA base number in the gene by adding the part of the DNA.
b) Deletion:
This type of mutation occurs when there is a difference in the number of DNA bases by eliminating a piece of DNA.
3. Base substitution Mutations:
This type of mutations occur when there is replacement of one base pair by the other base pair. This mutations are further classified as Transition mutation and transversion mutation,
a) Transition mutation: This occurs when the base of one chemical is replaced by the other base of the same chemical molecule (4). It mainly happens when there is the transposing of the purine molecules i.e., A is transposed by G or by the transposing of pyrimidine molecules i.e., C by T in the DNA molecule.
b) Tranvsersion Mutation: This occurs when there is an opposite replacement of a category base chemical by another base of the other category . This is mainly due to the incorrect replacement of the DNA bases i.e., when a pyrimidine is replaced with purine molecule.
A change in the genetic structure that is not inherited from a parent, and also not passed to offspring, is called a somatic mutation.[86] Somatic mutations are not inherited by an organism’s offspring because they do not affect the germline. However, they are passed down to all the progeny of a mutated cell within the same organism during mitosis. A major section of an organism therefore might carry the same mutation. These types of mutations are usually prompted by environmental causes, such as ultraviolet radiation or any exposure to certain harmful chemicals, and can cause diseases including cancer.[91]
With plants, some somatic mutations can be propagated without the need for seed production, for example, by grafting and stem cuttings. These type of mutation have led to new types of fruits, such as the «Delicious» apple and the «Washington» navel orange.[92]
Human and mouse somatic cells have a mutation rate more than ten times higher than the germline mutation rate for both species; mice have a higher rate of both somatic and germline mutations per cell division than humans. The disparity in mutation rate between the germline and somatic tissues likely reflects the greater importance of genome maintenance in the germline than in the soma.[93]
Special classes[edit]
- Conditional mutation is a mutation that has wild-type (or less severe) phenotype under certain «permissive» environmental conditions and a mutant phenotype under certain «restrictive» conditions. For example, a temperature-sensitive mutation can cause cell death at high temperature (restrictive condition), but might have no deleterious consequences at a lower temperature (permissive condition).[94] These mutations are non-autonomous, as their manifestation depends upon presence of certain conditions, as opposed to other mutations which appear autonomously.[95] The permissive conditions may be temperature,[96] certain chemicals,[97] light[97] or mutations in other parts of the genome.[95] In vivo mechanisms like transcriptional switches can create conditional mutations. For instance, association of Steroid Binding Domain can create a transcriptional switch that can change the expression of a gene based on the presence of a steroid ligand.[98] Conditional mutations have applications in research as they allow control over gene expression. This is especially useful studying diseases in adults by allowing expression after a certain period of growth, thus eliminating the deleterious effect of gene expression seen during stages of development in model organisms.[97] DNA Recombinase systems like Cre-Lox recombination used in association with promoters that are activated under certain conditions can generate conditional mutations. Dual Recombinase technology can be used to induce multiple conditional mutations to study the diseases which manifest as a result of simultaneous mutations in multiple genes.[97] Certain inteins have been identified which splice only at certain permissive temperatures, leading to improper protein synthesis and thus, loss-of-function mutations at other temperatures.[99] Conditional mutations may also be used in genetic studies associated with ageing, as the expression can be changed after a certain time period in the organism’s lifespan.[96]
- Replication timing quantitative trait loci affects DNA replication.
Nomenclature[edit]
In order to categorize a mutation as such, the «normal» sequence must be obtained from the DNA of a «normal» or «healthy» organism (as opposed to a «mutant» or «sick» one), it should be identified and reported; ideally, it should be made publicly available for a straightforward nucleotide-by-nucleotide comparison, and agreed upon by the scientific community or by a group of expert geneticists and biologists, who have the responsibility of establishing the standard or so-called «consensus» sequence. This step requires a tremendous scientific effort. Once the consensus sequence is known, the mutations in a genome can be pinpointed, described, and classified. The committee of the Human Genome Variation Society (HGVS) has developed the standard human sequence variant nomenclature,[100] which should be used by researchers and DNA diagnostic centers to generate unambiguous mutation descriptions. In principle, this nomenclature can also be used to describe mutations in other organisms. The nomenclature specifies the type of mutation and base or amino acid changes.
- Nucleotide substitution (e.g., 76A>T) – The number is the position of the nucleotide from the 5′ end; the first letter represents the wild-type nucleotide, and the second letter represents the nucleotide that replaced the wild type. In the given example, the adenine at the 76th position was replaced by a thymine.
- If it becomes necessary to differentiate between mutations in genomic DNA, mitochondrial DNA, and RNA, a simple convention is used. For example, if the 100th base of a nucleotide sequence mutated from G to C, then it would be written as g.100G>C if the mutation occurred in genomic DNA, m.100G>C if the mutation occurred in mitochondrial DNA, or r.100g>c if the mutation occurred in RNA. Note that, for mutations in RNA, the nucleotide code is written in lower case.
- Amino acid substitution (e.g., D111E) – The first letter is the one letter code of the wild-type amino acid, the number is the position of the amino acid from the N-terminus, and the second letter is the one letter code of the amino acid present in the mutation. Nonsense mutations are represented with an X for the second amino acid (e.g. D111X).
- Amino acid deletion (e.g., ΔF508) – The Greek letter Δ (delta) indicates a deletion. The letter refers to the amino acid present in the wild type and the number is the position from the N terminus of the amino acid were it to be present as in the wild type.
Mutation rates[edit]
Mutation rates vary substantially across species, and the evolutionary forces that generally determine mutation are the subject of ongoing investigation.
In humans, the mutation rate is about 50-90 de novo mutations per genome per generation, that is, each human accumulates about 50-90 novel mutations that were not present in his or her parents. This number has been established by sequencing thousands of human trios, that is, two parents and at least one child.[101]
The genomes of RNA viruses are based on RNA rather than DNA. The RNA viral genome can be double-stranded (as in DNA) or single-stranded. In some of these viruses (such as the single-stranded human immunodeficiency virus), replication occurs quickly, and there are no mechanisms to check the genome for accuracy. This error-prone process often results in mutations.
Randomness of mutations[edit]
There is a widespread assumption that mutations are (entirely) «random» with respect to their consequences (in terms of probability). This was shown to be wrong as mutation frequency can vary across regions of the genome, with such DNA repair- and mutation-biases being associated with various factors. For instance, biologically important regions were found to be protected from mutations and mutations beneficial to the studied plant were found to be more likely – i.e. mutation is «non-random in a way that benefits the plant».[102][103]
Disease causation[edit]
Changes in DNA caused by mutation in a coding region of DNA can cause errors in protein sequence that may result in partially or completely non-functional proteins. Each cell, in order to function correctly, depends on thousands of proteins to function in the right places at the right times. When a mutation alters a protein that plays a critical role in the body, a medical condition can result. One study on the comparison of genes between different species of Drosophila suggests that if a mutation does change a protein, the mutation will most likely be harmful, with an estimated 70 percent of amino acid polymorphisms having damaging effects, and the remainder being either neutral or weakly beneficial.[8] Some mutations alter a gene’s DNA base sequence but do not change the protein made by the gene. Studies have shown that only 7% of point mutations in noncoding DNA of yeast are deleterious and 12% in coding DNA are deleterious. The rest of the mutations are either neutral or slightly beneficial.[104]
Inherited disorders[edit]
If a mutation is present in a germ cell, it can give rise to offspring that carries the mutation in all of its cells. This is the case in hereditary diseases. In particular, if there is a mutation in a DNA repair gene within a germ cell, humans carrying such germline mutations may have an increased risk of cancer. A list of 34 such germline mutations is given in the article DNA repair-deficiency disorder. An example of one is albinism, a mutation that occurs in the OCA1 or OCA2 gene. Individuals with this disorder are more prone to many types of cancers, other disorders and have impaired vision.
DNA damage can cause an error when the DNA is replicated, and this error of replication can cause a gene mutation that, in turn, could cause a genetic disorder. DNA damages are repaired by the DNA repair system of the cell. Each cell has a number of pathways through which enzymes recognize and repair damages in DNA. Because DNA can be damaged in many ways, the process of DNA repair is an important way in which the body protects itself from disease. Once DNA damage has given rise to a mutation, the mutation cannot be repaired.
Role in carcinogenesis[edit]
On the other hand, a mutation may occur in a somatic cell of an organism. Such mutations will be present in all descendants of this cell within the same organism. The accumulation of certain mutations over generations of somatic cells is part of cause of malignant transformation, from normal cell to cancer cell.[105]
Cells with heterozygous loss-of-function mutations (one good copy of gene and one mutated copy) may function normally with the unmutated copy until the good copy has been spontaneously somatically mutated. This kind of mutation happens often in living organisms, but it is difficult to measure the rate. Measuring this rate is important in predicting the rate at which people may develop cancer.[106]
Point mutations may arise from spontaneous mutations that occur during DNA replication. The rate of mutation may be increased by mutagens. Mutagens can be physical, such as radiation from UV rays, X-rays or extreme heat, or chemical (molecules that misplace base pairs or disrupt the helical shape of DNA). Mutagens associated with cancers are often studied to learn about cancer and its prevention.
Prion mutations[edit]
Prions are proteins and do not contain genetic material. However, prion replication has been shown to be subject to mutation and natural selection just like other forms of replication.[107] The human gene PRNP codes for the major prion protein, PrP, and is subject to mutations that can give rise to disease-causing prions.
Beneficial mutations[edit]
Although mutations that cause changes in protein sequences can be harmful to an organism, on occasions the effect may be positive in a given environment. In this case, the mutation may enable the mutant organism to withstand particular environmental stresses better than wild-type organisms, or reproduce more quickly. In these cases a mutation will tend to become more common in a population through natural selection. Examples include the following:
HIV resistance: a specific 32 base pair deletion in human CCR5 (CCR5-Δ32) confers HIV resistance to homozygotes and delays AIDS onset in heterozygotes.[108] One possible explanation of the etiology of the relatively high frequency of CCR5-Δ32 in the European population is that it conferred resistance to the bubonic plague in mid-14th century Europe. People with this mutation were more likely to survive infection; thus its frequency in the population increased.[109] This theory could explain why this mutation is not found in Southern Africa, which remained untouched by bubonic plague. A newer theory suggests that the selective pressure on the CCR5 Delta 32 mutation was caused by smallpox instead of the bubonic plague.[110]
Malaria resistance: An example of a harmful mutation is sickle-cell disease, a blood disorder in which the body produces an abnormal type of the oxygen-carrying substance hemoglobin in the red blood cells. One-third of all indigenous inhabitants of Sub-Saharan Africa carry the allele, because, in areas where malaria is common, there is a survival value in carrying only a single sickle-cell allele (sickle cell trait).[111] Those with only one of the two alleles of the sickle-cell disease are more resistant to malaria, since the infestation of the malaria Plasmodium is halted by the sickling of the cells that it infests.
Antibiotic resistance: Practically all bacteria develop antibiotic resistance when exposed to antibiotics. In fact, bacterial populations already have such mutations that get selected under antibiotic selection.[112] Obviously, such mutations are only beneficial for the bacteria but not for those infected.
Lactase persistence. A mutation allowed humans to express the enzyme lactase after they are naturally weaned from breast milk, allowing adults to digest lactose, which is likely one of the most beneficial mutations in recent human evolution.[113]
Compensated pathogenic deviations[edit]
Compensated pathogenic deviations refer to amino acid residues in a protein sequence that are pathogenic in one species but are wild type residues in the functionally equivalent protein in another species. Although the amino acid residue is pathogenic in the first species, it is not so in the second species because its pathogenicity is compensated by one or more amino acid substitutions in the second species. The compensatory mutation can occur in the same protein or in another protein with which it interacts.[114]
It is critical to understand the effects of compensatory mutations in the context of fixed deleterious mutations due to the population fitness decreasing because of fixation.[115] Effective population size refers to a population that is reproducing.[116] An increase in this population size has been correlated with a decreased rate of genetic diversity.[116] The position of a population relative to the critical effect population size is essential to determine the effect deleterious alleles will have on fitness.[115] If the population is below the critical effective size fitness will decrease drastically, however if the population is above the critical effect size, fitness can increase regardless of deleterious mutations due to compensatory alleles.[115]
Compensatory mutations in RNA[edit]
As the function of a RNA molecule is dependent on its structure,[117] the structure of RNA molecules is evolutionarily conserved. Therefore, any mutation that alters the stable structure of RNA molecules must be compensated by other compensatory mutations. In the context of RNA, the sequence of the RNA can be considered as ‘ genotype’ and the structure of the RNA can be considered as its ‘phenotype’. Since RNAs have relatively simpler composition than proteins, the structure of RNA molecules can be computationally predicted with high degree of accuracy. Because of this convenience, compensatory mutations have been studied in computational simulations using RNA folding algorithms.[118][119]
Evolutionary mechanism of compensation[edit]
Compensatory mutations can be explained by the genetic phenomenon epistasis whereby the phenotypic effect of one mutation is dependent upon mutation(s) at other loci. While epistasis was originally conceived in the context of interaction between different genes, intragenic epistasis has also been studied recently.[120] Existence of compensated pathogenic deviations can be explained by ‘sign epistasis’, in which the effects of a deleterious mutation can be compensated by the presence of a epistatic mutation in another loci. For a given protein, a deleterious mutation (D) and a compensatory mutation (C) can be considered, where C can be in the same protein as D or in a different interacting protein depending on the context. The fitness effect of C itself could be neutral or somewhat deleterious such that it can still exist in the population, and the effect of D is deleterious to the extent that it cannot exist in the population. However, when C and D co-occur together, the combined fitness effect becomes neutral or positive.[114] Thus, compensatory mutations can bring novelty to proteins by forging new pathways of protein evolution : it allows individuals to travel from one fitness peak to another through the valleys of lower fitness.[120]
DePristo et al. 2005 outlined two models to explain the dynamics of compensatory pathogenic deviations (CPD).[121] In the first hypothesis P is a pathogenic amino acid mutation that and C is a neutral compensatory mutation.[121] Under these conditions, if the pathogenic mutation arises after a compensatory mutation, then P can become fixed in the population.[121] The second model of CPDs states that P and C are both deleterious mutations resulting in fitness valleys when mutations occur simultaneously.[121] Using publicly available, Ferrer-Costa et al. 2007 obtained compensatory mutations and human pathogenic mutation datasets that were characterized to determine what causes CPDs.[122] Results indicate that the structural constraints and the location in protein structure determine whether compensated mutations will occur.[122]
Experimental evidence of compensatory mutations[edit]
Experiment in bacteria[edit]
Lunzer et al.[123] tested the outcome of swapping divergent amino acids between two orthologous proteins of isopropymalate dehydrogenase (IMDH). They substituted 168 amino acids in Escherichia coli IMDH that are wild type residues in IMDH Pseudomonas aeruginosa. They found that over one third of these substitutions compromised IMDH enzymatic activity in the Escherichia coli genetic background. This demonstrated that identical amino acid states can result in different phenotypic states depending on the genetic background. Corrigan et al. 2011 demonstrated how staphylococcus aureus was able to grow normally without the presence of lipoteichoic acid due to compensatory mutations.[124] Whole genome sequencing results revealed that when Cyclic-di-AMP phosphodiesterase (GdpP) was disrupted in this bacterium, it compensated for the disappearance of the cell wall polymer, resulting in normal cell growth.[124]
Research has shown that bacteria can gain drug resistance through compensatory mutations that do not impede or having little effect on fitness.[125] Previous research from Gagneux et al. 2006 has found that laboratory grown M. tuberculosis strains with rifampicin resistance have reduced fitness, however drug resistant clinical strains of this pathogenic bacteria do not have reduced fitness.[126] Comas et al. 2012 used whole genome comparisons between clinical strains and lab derived mutants to determine the role and contribution of compensatory mutations in drug resistance to rifampicin.[125] Genome analysis reveal rifampicin resistant strains have a mutation in rpoA and rpoC.[125] A similar study investigated the bacterial fitness associated with compensatory mutations in rifampin resistant Escherichia coli.[127] Results obtained from this study demonstrate that drug resistance is linked to bacterial fitness as higher fitness costs are linked to greater transcription errors.[127]
Experiment in virus[edit]
Gong et al.[128] collected obtained genotype data of influenza nucleoprotein from different timelines and temporally ordered them according to their time of origin. Then they isolated 39 amino acid substitutions that occurred in different timelines and substituted them in a genetic background that approximated the ancestral genotype. They found that 3 of the 39 substitutions significantly reduced the fitness of the ancestral background. Compensatory mutations are new mutations that arise and have a positive or neutral impact on a populations fitness.[129] Previous research has shown that populations have can compensate detrimental mutations.[114][129][130] Burch and Chao tested Fisher’s geometric model of adaptive evolution by testing whether bacteriophage φ6 evolves by small steps.[131] Their results showed that bacteriophage φ6 fitness declined rapidly and recovered in small steps .[131] Viral nucleoproteins have been shown to avoid cytotoxic T lymphocytes (CTLs) through arginine-to glycine substitutions.[132] This substitution mutations impacts the fitness of viral nucleoproteins, however compensatory co-mutations impede fitness declines and aid the virus to avoid recognition from CTLs.[132] Mutations can have three different effects; mutations can have deleterious effects, some increase fitness through compensatory mutations, and lastly mutations can be counterbalancing resulting in compensatory neutral mutations.[133][127][126]
History[edit]

Mutationism is one of several alternatives to Darwinian evolution that have existed both before and after the publication of Charles Darwin’s 1859 book, On the Origin of Species. In the theory, mutation was the source of novelty, creating new forms and new species, potentially instantaneously,[134] in a sudden jump.[135] This was envisaged as driving evolution, which was limited by the supply of mutations.
Before Darwin, biologists commonly believed in saltationism, the possibility of large evolutionary jumps, including immediate speciation. For example, in 1822 Étienne Geoffroy Saint-Hilaire argued that species could be formed by sudden transformations, or what would later be called macromutation.[136] Darwin opposed saltation, insisting on gradualism in evolution as in geology. In 1864, Albert von Kölliker revived Geoffroy’s theory.[137] In 1901 the geneticist Hugo de Vries gave the name «mutation» to seemingly new forms that suddenly arose in his experiments on the evening primrose Oenothera lamarckiana, and in the first decade of the 20th century, mutationism, or as de Vries named it mutationstheorie,[134][138] became a rival to Darwinism supported for a while by geneticists including William Bateson,[139] Thomas Hunt Morgan, and Reginald Punnett.[134][140]
Understanding of mutationism is clouded by the mid-20th century portrayal of the early mutationists by supporters of the modern synthesis as opponents of Darwinian evolution and rivals of the biometrics school who argued that selection operated on continuous variation. In this portrayal, mutationism was defeated by a synthesis of genetics and natural selection that supposedly started later, around 1918, with work by the mathematician Ronald Fisher.[141][142][143][144] However, the alignment of Mendelian genetics and natural selection began as early as 1902 with a paper by Udny Yule,[145] and built up with theoretical and experimental work in Europe and America. Despite the controversy, the early mutationists had by 1918 already accepted natural selection and explained continuous variation as the result of multiple genes acting on the same characteristic, such as height.[142][143]
Mutationism, along with other alternatives to Darwinism like Lamarckism and orthogenesis, was discarded by most biologists as they came to see that Mendelian genetics and natural selection could readily work together; mutation took its place as a source of the genetic variation essential for natural selection to work on. However, mutationism did not entirely vanish. In 1940, Richard Goldschmidt again argued for single-step speciation by macromutation, describing the organisms thus produced as «hopeful monsters», earning widespread ridicule.[146][147] In 1987, Masatoshi Nei argued controversially that evolution was often mutation-limited.[148] Modern biologists such as Douglas J. Futuyma conclude that essentially all claims of evolution driven by large mutations can be explained by Darwinian evolution.[149]
See also[edit]
- Aneuploidy
- Antioxidant
- Budgerigar colour genetics
- DbDNV (2010)
- Deletion (genetics)
- Ecogenetics
- Embryology
- Homeobox
- Human somatic variation
- Polyploidy
- Robertsonian translocation
- Signature-tagged mutagenesis
- Somatic hypermutation
- TILLING (molecular biology)
- Trinucleotide repeat expansion
References[edit]
- ^ «mutation | Learn Science at Scitable». Nature. Nature Education. Retrieved 24 September 2018.
- ^ Sfeir A, Symington LS (November 2015). «Microhomology-Mediated End Joining: A Back-up Survival Mechanism or Dedicated Pathway?». Trends in Biochemical Sciences. 40 (11): 701–714. doi:10.1016/j.tibs.2015.08.006. PMC 4638128. PMID 26439531.
- ^ Chen J, Miller BF, Furano AV (April 2014). «Repair of naturally occurring mismatches can induce mutations in flanking DNA». eLife. 3: e02001. doi:10.7554/elife.02001. PMC 3999860. PMID 24843013.
- ^ Rodgers K, McVey M (January 2016). «Error-Prone Repair of DNA Double-Strand Breaks». Journal of Cellular Physiology. 231 (1): 15–24. doi:10.1002/jcp.25053. PMC 4586358. PMID 26033759.
- ^ a b Bertram JS (December 2000). «The molecular biology of cancer». Molecular Aspects of Medicine. 21 (6): 167–223. doi:10.1016/S0098-2997(00)00007-8. PMID 11173079. S2CID 24155688.
- ^ a b Aminetzach YT, Macpherson JM, Petrov DA (July 2005). «Pesticide resistance via transposition-mediated adaptive gene truncation in Drosophila». Science. 309 (5735): 764–7. Bibcode:2005Sci…309..764A. doi:10.1126/science.1112699. PMID 16051794. S2CID 11640993.
- ^ Burrus V, Waldor MK (June 2004). «Shaping bacterial genomes with integrative and conjugative elements». Research in Microbiology. 155 (5): 376–86. doi:10.1016/j.resmic.2004.01.012. PMID 15207870.
- ^ a b Sawyer SA, Parsch J, Zhang Z, Hartl DL (April 2007). «Prevalence of positive selection among nearly neutral amino acid replacements in Drosophila». Proceedings of the National Academy of Sciences of the United States of America. 104 (16): 6504–10. Bibcode:2007PNAS..104.6504S. doi:10.1073/pnas.0701572104. PMC 1871816. PMID 17409186.
- ^ Hastings PJ, Lupski JR, Rosenberg SM, Ira G (August 2009). «Mechanisms of change in gene copy number». Nature Reviews. Genetics. 10 (8): 551–64. doi:10.1038/nrg2593. PMC 2864001. PMID 19597530.
- ^ Carroll SB, Grenier JK, Weatherbee SD (2005). From DNA to Diversity: Molecular Genetics and the Evolution of Animal Design (2nd ed.). Malden, MA: Blackwell Publishing. ISBN 978-1-4051-1950-4. LCCN 2003027991. OCLC 53972564.
- ^ Harrison PM, Gerstein M (May 2002). «Studying genomes through the aeons: protein families, pseudogenes and proteome evolution». Journal of Molecular Biology. 318 (5): 1155–74. doi:10.1016/S0022-2836(02)00109-2. PMID 12083509.
- ^ Orengo CA, Thornton JM (July 2005). «Protein families and their evolution-a structural perspective». Annual Review of Biochemistry. 74: 867–900. doi:10.1146/annurev.biochem.74.082803.133029. PMID 15954844.
- ^ Long M, Betrán E, Thornton K, Wang W (November 2003). «The origin of new genes: glimpses from the young and old». Nature Reviews. Genetics. 4 (11): 865–75. doi:10.1038/nrg1204. PMID 14634634. S2CID 33999892.
- ^ Wang M, Caetano-Anollés G (January 2009). «The evolutionary mechanics of domain organization in proteomes and the rise of modularity in the protein world». Structure. 17 (1): 66–78. doi:10.1016/j.str.2008.11.008. PMID 19141283.
- ^ Bowmaker JK (May 1998). «Evolution of colour vision in vertebrates». Eye. 12 (Pt 3b): 541–7. doi:10.1038/eye.1998.143. PMID 9775215. S2CID 12851209.
- ^ Gregory TR, Hebert PD (April 1999). «The modulation of DNA content: proximate causes and ultimate consequences». Genome Research. 9 (4): 317–24. doi:10.1101/gr.9.4.317. PMID 10207154. S2CID 16791399.
- ^ Hurles M (July 2004). «Gene duplication: the genomic trade in spare parts». PLOS Biology. 2 (7): E206. doi:10.1371/journal.pbio.0020206. PMC 449868. PMID 15252449.
- ^ Liu N, Okamura K, Tyler DM, Phillips MD, Chung WJ, Lai EC (October 2008). «The evolution and functional diversification of animal microRNA genes». Cell Research. 18 (10): 985–96. doi:10.1038/cr.2008.278. PMC 2712117. PMID 18711447.
- ^ Siepel A (October 2009). «Darwinian alchemy: Human genes from noncoding DNA». Genome Research. 19 (10): 1693–5. doi:10.1101/gr.098376.109. PMC 2765273. PMID 19797681.
- ^ Zhang J, Wang X, Podlaha O (May 2004). «Testing the chromosomal speciation hypothesis for humans and chimpanzees». Genome Research. 14 (5): 845–51. doi:10.1101/gr.1891104. PMC 479111. PMID 15123584.
- ^ Ayala FJ, Coluzzi M (May 2005). «Chromosome speciation: humans, Drosophila, and mosquitoes». Proceedings of the National Academy of Sciences of the United States of America. 102 (Suppl 1): 6535–42. Bibcode:2005PNAS..102.6535A. doi:10.1073/pnas.0501847102. PMC 1131864. PMID 15851677.
- ^ Hurst GD, Werren JH (August 2001). «The role of selfish genetic elements in eukaryotic evolution». Nature Reviews Genetics. 2 (8): 597–606. doi:10.1038/35084545. PMID 11483984. S2CID 2715605.
- ^ Häsler J, Strub K (November 2006). «Alu elements as regulators of gene expression». Nucleic Acids Research. 34 (19): 5491–7. doi:10.1093/nar/gkl706. PMC 1636486. PMID 17020921.
- ^ a b c d Eyre-Walker A, Keightley PD (August 2007). «The distribution of fitness effects of new mutations» (PDF). Nature Reviews Genetics. 8 (8): 610–8. doi:10.1038/nrg2146. PMID 17637733. S2CID 10868777. Archived from the original (PDF) on 4 March 2016. Retrieved 6 September 2010.
- ^ a b Kimura M (1983). The Neutral Theory of Molecular Evolution. Cambridge, UK; New York: Cambridge University Press. ISBN 978-0-521-23109-1. LCCN 82022225. OCLC 9081989.
- ^ Bohidar HB (January 2015). Fundamentals of Polymer Physics and Molecular Biophysics. Cambridge University Press. ISBN 978-1-316-09302-3.
- ^ Dover GA, Darwin C (2000). Dear Mr. Darwin: Letters on the Evolution of Life and Human Nature. University of California Press. ISBN 9780520227903.
- ^ Tibayrenc M (12 January 2017). Genetics and Evolution of Infectious Diseases. Elsevier. ISBN 9780128001530.
- ^ «Cancer Is Partly Caused By Bad Luck, Study Finds». NPR.org. Archived from the original on 13 July 2017.
- ^ Jha A (22 August 2012). «Older fathers pass on more genetic mutations, study shows». The Guardian.
- ^ Ames BN, Shigenaga MK, Hagen TM (September 1993). «Oxidants, antioxidants, and the degenerative diseases of aging». Proceedings of the National Academy of Sciences of the United States of America. 90 (17): 7915–22. Bibcode:1993PNAS…90.7915A. doi:10.1073/pnas.90.17.7915. PMC 47258. PMID 8367443.
- ^ Montelone BA (1998). «Mutation, Mutagens, and DNA Repair». www-personal.ksu.edu. Archived from the original on 26 September 2015. Retrieved 2 October 2015.
- ^ Slocombe L, Al-Khalili JS, Sacchi M (February 2021). «Quantum and classical effects in DNA point mutations: Watson-Crick tautomerism in AT and GC base pairs». Physical Chemistry Chemical Physics. 23 (7): 4141–4150. Bibcode:2021PCCP…23.4141S. doi:10.1039/D0CP05781A. ISSN 1463-9076. PMID 33533770. S2CID 231788542.
- ^ Slocombe L, Sacchi M, Al-Khalili J (5 May 2022). «An open quantum systems approach to proton tunnelling in DNA». Communications Physics. 5 (1): 109. arXiv:2110.00113. Bibcode:2022CmPhy…5..109S. doi:10.1038/s42005-022-00881-8. ISSN 2399-3650. S2CID 238253421.
- ^ Stuart GR, Oda Y, de Boer JG, Glickman BW (March 2000). «Mutation frequency and specificity with age in liver, bladder and brain of lacI transgenic mice». Genetics. 154 (3): 1291–300. doi:10.1093/genetics/154.3.1291. PMC 1460990. PMID 10757770.
- ^ Kunz BA, Ramachandran K, Vonarx EJ (April 1998). «DNA sequence analysis of spontaneous mutagenesis in Saccharomyces cerevisiae». Genetics. 148 (4): 1491–505. doi:10.1093/genetics/148.4.1491. PMC 1460101. PMID 9560369.
- ^ Lieber MR (July 2010). «The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway». Annual Review of Biochemistry. 79: 181–211. doi:10.1146/annurev.biochem.052308.093131. PMC 3079308. PMID 20192759.
- ^ Created from PDB 1JDG Archived 31 December 2015 at the Wayback Machine
- ^ Pfohl-Leszkowicz A, Manderville RA (January 2007). «Ochratoxin A: An overview on toxicity and carcinogenicity in animals and humans». Molecular Nutrition & Food Research. 51 (1): 61–99. doi:10.1002/mnfr.200600137. PMID 17195275.
- ^ Kozmin S, Slezak G, Reynaud-Angelin A, Elie C, de Rycke Y, Boiteux S, Sage E (September 2005). «UVA radiation is highly mutagenic in cells that are unable to repair 7,8-dihydro-8-oxoguanine in Saccharomyces cerevisiae». Proceedings of the National Academy of Sciences of the United States of America. 102 (38): 13538–43. Bibcode:2005PNAS..10213538K. doi:10.1073/pnas.0504497102. PMC 1224634. PMID 16157879.
- ^ a b Fitzgerald DM, Rosenberg SM (April 2019). «What is mutation? A chapter in the series: How microbes «jeopardize» the modern synthesis». PLOS Genetics. 15 (4): e1007995. doi:10.1371/journal.pgen.1007995. PMC 6443146. PMID 30933985.
- ^ Galhardo RS, Hastings PJ, Rosenberg SM (1 January 2007). «Mutation as a stress response and the regulation of evolvability». Critical Reviews in Biochemistry and Molecular Biology. 42 (5): 399–435. doi:10.1080/10409230701648502. PMC 3319127. PMID 17917874.
- ^ Quinto-Alemany D, Canerina-Amaro A, Hernández-Abad LG, Machín F, Romesberg FE, Gil-Lamaignere C (31 July 2012). Sturtevant J (ed.). «Yeasts acquire resistance secondary to antifungal drug treatment by adaptive mutagenesis». PLOS ONE. 7 (7): e42279. Bibcode:2012PLoSO…742279Q. doi:10.1371/journal.pone.0042279. PMC 3409178. PMID 22860105.
- ^ Rahman N. «The clinical impact of DNA sequence changes». Transforming Genetic Medicine Initiative. Archived from the original on 4 August 2017. Retrieved 27 June 2017.
- ^ Freese E (April 1959). «The Difference Between Spontaneous and Base-Analogue Induced Mutations of Phage T4». Proceedings of the National Academy of Sciences of the United States of America. 45 (4): 622–33. Bibcode:1959PNAS…45..622F. doi:10.1073/pnas.45.4.622. PMC 222607. PMID 16590424.
- ^ Freese E (June 1959). «The specific mutagenic effect of base analogues on Phage T4». Journal of Molecular Biology. 1 (2): 87–105. doi:10.1016/S0022-2836(59)80038-3.
- ^ References for the image are found in Wikimedia Commons page at: Commons:File:Notable mutations.svg#References.
- ^ Hogan CM (12 October 2010). «Mutation». In Monosson E (ed.). Encyclopedia of Earth. Washington, D.C.: Environmental Information Coalition, National Council for Science and the Environment. OCLC 72808636. Archived from the original on 14 November 2015. Retrieved 8 October 2015.
- ^ Boillée S, Vande Velde C, Cleveland DW (October 2006). «ALS: a disease of motor neurons and their nonneuronal neighbors». Neuron. 52 (1): 39–59. CiteSeerX 10.1.1.325.7514. doi:10.1016/j.neuron.2006.09.018. PMID 17015226. S2CID 12968143.
- ^ Reva B, Antipin Y, Sander C (September 2011). «Predicting the functional impact of protein mutations: application to cancer genomics». Nucleic Acids Research. 39 (17): e118. doi:10.1093/nar/gkr407. PMC 3177186. PMID 21727090.
- ^ Housden BE, Muhar M, Gemberling M, Gersbach CA, Stainier DY, Seydoux G, et al. (January 2017). «Loss-of-function genetic tools for animal models: cross-species and cross-platform differences». Nature Reviews. Genetics. 18 (1): 24–40. doi:10.1038/nrg.2016.118. PMC 5206767. PMID 27795562.
- ^ Board on Life Sciences; Division on Earth and Life Studies; Committee on Science Technology; Policy and Global Affairs; Board on Health Sciences Policy; National Research Council; Institute of Medicine (13 April 2015). Gain-of-Function Research: Background and Alternatives. National Academies Press (US).
- ^ Eggertsson G, Adelberg EA (August 1965). «Map positions and specificities of suppressor mutations in Escherichia coli K-12». Genetics. 52 (2): 319–340. doi:10.1093/genetics/52.2.319. PMC 1210853. PMID 5324068.
- ^ Takiar V, Ip CK, Gao M, Mills GB, Cheung LW (March 2017). «Neomorphic mutations create therapeutic challenges in cancer». Oncogene. 36 (12): 1607–1618. doi:10.1038/onc.2016.312. PMC 6609160. PMID 27841866.
- ^ Ellis NA, Ciocci S, German J (February 2001). «Back mutation can produce phenotype reversion in Bloom syndrome somatic cells». Human Genetics. 108 (2): 167–73. doi:10.1007/s004390000447. PMID 11281456. S2CID 22290041.
- ^ Doolittle WF, Brunet TD (December 2017). «On causal roles and selected effects: our genome is mostly junk». BMC Biology. 15 (1): 116. doi:10.1186/s12915-017-0460-9. PMC 5718017. PMID 29207982.
- ^ Nichols RJ, Sen S, Choo YJ, Beltrao P, Zietek M, Chaba R, et al. (January 2011). «Phenotypic landscape of a bacterial cell». Cell. 144 (1): 143–56. doi:10.1016/j.cell.2010.11.052. PMC 3060659. PMID 21185072.
- ^ van Opijnen T, Bodi KL, Camilli A (October 2009). «Tn-seq: high-throughput parallel sequencing for fitness and genetic interaction studies in microorganisms». Nature Methods. 6 (10): 767–72. doi:10.1038/nmeth.1377. PMC 2957483. PMID 19767758.
- ^ Allen HL, Estrada K, Lettre G, Berndt SI, Weedon MN, Rivadeneira F, et al. (October 2010). «Hundreds of variants clustered in genomic loci and biological pathways affect human height». Nature. 467 (7317): 832–8. Bibcode:2010Natur.467..832L. doi:10.1038/nature09410. PMC 2955183. PMID 20881960.
- ^ Charlesworth D, Charlesworth B, Morgan MT (December 1995). «The pattern of neutral molecular variation under the background selection model». Genetics. 141 (4): 1619–32. doi:10.1093/genetics/141.4.1619. PMC 1206892. PMID 8601499.
- ^ Loewe L (April 2006). «Quantifying the genomic decay paradox due to Muller’s ratchet in human mitochondrial DNA». Genetical Research. 87 (2): 133–59. doi:10.1017/S0016672306008123. PMID 16709275.
- ^ Bernstein H, Hopf FA, Michod RE (1987). «The molecular basis of the evolution of sex». Molecular Genetics of Development. Advances in Genetics. Vol. 24. pp. 323–70. doi:10.1016/s0065-2660(08)60012-7. ISBN 9780120176243. PMID 3324702.
- ^ Peck JR, Barreau G, Heath SC (April 1997). «Imperfect genes, Fisherian mutation and the evolution of sex». Genetics. 145 (4): 1171–99. doi:10.1093/genetics/145.4.1171. PMC 1207886. PMID 9093868.
- ^ Simcikova D, Heneberg P (December 2019). «Refinement of evolutionary medicine predictions based on clinical evidence for the manifestations of Mendelian diseases». Scientific Reports. 9 (1): 18577. Bibcode:2019NatSR…918577S. doi:10.1038/s41598-019-54976-4. PMC 6901466. PMID 31819097.
- ^ Keightley PD, Lynch M (March 2003). «Toward a realistic model of mutations affecting fitness». Evolution; International Journal of Organic Evolution. 57 (3): 683–5, discussion 686–9. doi:10.1554/0014-3820(2003)057[0683:tarmom]2.0.co;2. JSTOR 3094781. PMID 12703958. S2CID 198157678.
- ^ Barton NH, Keightley PD (January 2002). «Understanding quantitative genetic variation». Nature Reviews Genetics. 3 (1): 11–21. doi:10.1038/nrg700. PMID 11823787. S2CID 8934412.
- ^ a b c Sanjuán R, Moya A, Elena SF (June 2004). «The distribution of fitness effects caused by single-nucleotide substitutions in an RNA virus». Proceedings of the National Academy of Sciences of the United States of America. 101 (22): 8396–401. Bibcode:2004PNAS..101.8396S. doi:10.1073/pnas.0400146101. PMC 420405. PMID 15159545.
- ^ Carrasco P, de la Iglesia F, Elena SF (December 2007). «Distribution of fitness and virulence effects caused by single-nucleotide substitutions in Tobacco Etch virus». Journal of Virology. 81 (23): 12979–84. doi:10.1128/JVI.00524-07. PMC 2169111. PMID 17898073.
- ^ Sanjuán R (June 2010). «Mutational fitness effects in RNA and single-stranded DNA viruses: common patterns revealed by site-directed mutagenesis studies». Philosophical Transactions of the Royal Society of London. Series B, Biological Sciences. 365 (1548): 1975–82. doi:10.1098/rstb.2010.0063. PMC 2880115. PMID 20478892.
- ^ Peris JB, Davis P, Cuevas JM, Nebot MR, Sanjuán R (June 2010). «Distribution of fitness effects caused by single-nucleotide substitutions in bacteriophage f1». Genetics. 185 (2): 603–9. doi:10.1534/genetics.110.115162. PMC 2881140. PMID 20382832.
- ^ Elena SF, Ekunwe L, Hajela N, Oden SA, Lenski RE (March 1998). «Distribution of fitness effects caused by random insertion mutations in Escherichia coli». Genetica. 102–103 (1–6): 349–58. doi:10.1023/A:1017031008316. PMID 9720287. S2CID 2267064.
- ^ a b Hietpas RT, Jensen JD, Bolon DN (May 2011). «Experimental illumination of a fitness landscape». Proceedings of the National Academy of Sciences of the United States of America. 108 (19): 7896–901. Bibcode:2011PNAS..108.7896H. doi:10.1073/pnas.1016024108. PMC 3093508. PMID 21464309.
- ^ Davies EK, Peters AD, Keightley PD (September 1999). «High frequency of cryptic deleterious mutations in Caenorhabditis elegans». Science. 285 (5434): 1748–51. doi:10.1126/science.285.5434.1748. PMID 10481013.
- ^ Loewe L, Charlesworth B (September 2006). «Inferring the distribution of mutational effects on fitness in Drosophila». Biology Letters. 2 (3): 426–30. doi:10.1098/rsbl.2006.0481. PMC 1686194. PMID 17148422.
- ^ Eyre-Walker A, Woolfit M, Phelps T (June 2006). «The distribution of fitness effects of new deleterious amino acid mutations in humans». Genetics. 173 (2): 891–900. doi:10.1534/genetics.106.057570. PMC 1526495. PMID 16547091.
- ^ Sawyer SA, Kulathinal RJ, Bustamante CD, Hartl DL (August 2003). «Bayesian analysis suggests that most amino acid replacements in Drosophila are driven by positive selection». Journal of Molecular Evolution. 57 (1): S154–64. Bibcode:2003JMolE..57S.154S. CiteSeerX 10.1.1.78.65. doi:10.1007/s00239-003-0022-3. PMID 15008412. S2CID 18051307.
- ^ Piganeau G, Eyre-Walker A (September 2003). «Estimating the distribution of fitness effects from DNA sequence data: implications for the molecular clock». Proceedings of the National Academy of Sciences of the United States of America. 100 (18): 10335–40. Bibcode:2003PNAS..10010335P. doi:10.1073/pnas.1833064100. PMC 193562. PMID 12925735.
- ^ Kimura M (February 1968). «Evolutionary rate at the molecular level». Nature. 217 (5129): 624–6. Bibcode:1968Natur.217..624K. doi:10.1038/217624a0. PMID 5637732. S2CID 4161261.
- ^ Akashi H (September 1999). «Within- and between-species DNA sequence variation and the ‘footprint’ of natural selection». Gene. 238 (1): 39–51. doi:10.1016/S0378-1119(99)00294-2. PMID 10570982.
- ^ Eyre-Walker A (October 2006). «The genomic rate of adaptive evolution». Trends in Ecology & Evolution. 21 (10): 569–75. doi:10.1016/j.tree.2006.06.015. PMID 16820244.
- ^ Gillespie JH (September 1984). «Molecular Evolution Over the Mutational Landscape». Evolution. 38 (5): 1116–1129. doi:10.2307/2408444. JSTOR 2408444. PMID 28555784.
- ^ Orr HA (April 2003). «The distribution of fitness effects among beneficial mutations». Genetics. 163 (4): 1519–26. doi:10.1093/genetics/163.4.1519. PMC 1462510. PMID 12702694.
- ^ Kassen R, Bataillon T (April 2006). «Distribution of fitness effects among beneficial mutations before selection in experimental populations of bacteria». Nature Genetics. 38 (4): 484–8. doi:10.1038/ng1751. PMID 16550173. S2CID 6954765.
- ^ Rokyta DR, Joyce P, Caudle SB, Wichman HA (April 2005). «An empirical test of the mutational landscape model of adaptation using a single-stranded DNA virus». Nature Genetics. 37 (4): 441–4. doi:10.1038/ng1535. PMID 15778707. S2CID 20296781.
- ^ Imhof M, Schlotterer C (January 2001). «Fitness effects of advantageous mutations in evolving Escherichia coli populations». Proceedings of the National Academy of Sciences of the United States of America. 98 (3): 1113–7. Bibcode:2001PNAS…98.1113I. doi:10.1073/pnas.98.3.1113. PMC 14717. PMID 11158603.
- ^ a b «Somatic cell genetic mutation». Genome Dictionary. Athens, Greece: Information Technology Associates. 30 June 2007. Archived from the original on 24 February 2010. Retrieved 6 June 2010.
- ^ «Compound heterozygote». MedTerms. New York: WebMD. 14 June 2012. Archived from the original on 4 March 2016. Retrieved 9 October 2015.
- ^ «RB1 Genetics». Daisy’s Eye Cancer Fund. Oxford, UK. Archived from the original on 26 November 2011. Retrieved 9 October 2015.
- ^ Cotton, RG (1993). «Current methods of mutation detection». Mutat Res. 285 (1): 125–144. doi:10.1016/0027-5107(93)90060-S. PMID 7678126.
- ^ a b Lerman, LS; Silverstein, K (1987). «Computational simulation of DNA melting and its application to denaturing gradient gel electrophoresis». Methods Enzymol. 155: 482–501. doi:10.1016/0076-6879(87)55032-7. PMID 2828875.
- ^ «somatic mutation | genetics». Encyclopædia Britannica. Archived from the original on 31 March 2017. Retrieved 31 March 2017.
- ^ Hartl L, Jones EW (1998). Genetics Principles and Analysis. Sudbury, Massachusetts: Jones and Bartlett Publishers. pp. 556. ISBN 978-0-7637-0489-6.
- ^ Milholland B, Dong X, Zhang L, Hao X, Suh Y, Vijg J (May 2017). «Differences between germline and somatic mutation rates in humans and mice». Nature Communications. 8: 15183. Bibcode:2017NatCo…815183M. doi:10.1038/ncomms15183. PMC 5436103. PMID 28485371.
- ^ Alberts B (2014). Molecular Biology of the Cell (6 ed.). Garland Science. p. 487. ISBN 9780815344322.
- ^ a b Chadov BF, Fedorova NB, Chadova EV (1 July 2015). «Conditional mutations in Drosophila melanogaster: On the occasion of the 150th anniversary of G. Mendel’s report in Brünn». Mutation Research/Reviews in Mutation Research. 765: 40–55. doi:10.1016/j.mrrev.2015.06.001. PMID 26281767.
- ^ a b Landis G, Bhole D, Lu L, Tower J (July 2001). «High-frequency generation of conditional mutations affecting Drosophila melanogaster development and life span». Genetics. 158 (3): 1167–76. doi:10.1093/genetics/158.3.1167. PMC 1461716. PMID 11454765. Archived from the original on 22 March 2017. Retrieved 21 March 2017.
- ^ a b c d Gierut JJ, Jacks TE, Haigis KM (April 2014). «Strategies to achieve conditional gene mutation in mice». Cold Spring Harbor Protocols. 2014 (4): 339–49. doi:10.1101/pdb.top069807. PMC 4142476. PMID 24692485.
- ^ Spencer DM (May 1996). «Creating conditional mutations in mammals». Trends in Genetics. 12 (5): 181–7. doi:10.1016/0168-9525(96)10013-5. PMID 8984733.
- ^ Tan G, Chen M, Foote C, Tan C (September 2009). «Temperature-sensitive mutations made easy: generating conditional mutations by using temperature-sensitive inteins that function within different temperature ranges». Genetics. 183 (1): 13–22. doi:10.1534/genetics.109.104794. PMC 2746138. PMID 19596904.
- ^ den Dunnen JT, Antonarakis SE (January 2000). «Mutation nomenclature extensions and suggestions to describe complex mutations: a discussion». Human Mutation. 15 (1): 7–12. doi:10.1002/(SICI)1098-1004(200001)15:1<7::AID-HUMU4>3.0.CO;2-N. PMID 10612815. S2CID 84706224.
- ^ Jónsson H, Sulem P, Kehr B, Kristmundsdottir S, Zink F, Hjartarson E, et al. (September 2017). «Parental influence on human germline de novo mutations in 1,548 trios from Iceland». Nature. 549 (7673): 519–522. Bibcode:2017Natur.549..519J. doi:10.1038/nature24018. PMID 28959963. S2CID 205260431.
- ^ «Study challenges evolutionary theory that DNA mutations are random». U.C. Davis. Retrieved 12 February 2022.
- ^ Monroe JG, Srikant T, Carbonell-Bejerano P, Becker C, Lensink M, Exposito-Alonso M, et al. (February 2022). «Mutation bias reflects natural selection in Arabidopsis thaliana». Nature. 602 (7895): 101–105. Bibcode:2022Natur.602..101M. doi:10.1038/s41586-021-04269-6. PMC 8810380. PMID 35022609.
- ^ Doniger SW, Kim HS, Swain D, Corcuera D, Williams M, Yang SP, Fay JC (August 2008). Pritchard JK (ed.). «A catalog of neutral and deleterious polymorphism in yeast». PLOS Genetics. 4 (8): e1000183. doi:10.1371/journal.pgen.1000183. PMC 2515631. PMID 18769710.
- ^ Ionov Y, Peinado MA, Malkhosyan S, Shibata D, Perucho M (June 1993). «Ubiquitous somatic mutations in simple repeated sequences reveal a new mechanism for colonic carcinogenesis». Nature. 363 (6429): 558–61. Bibcode:1993Natur.363..558I. doi:10.1038/363558a0. PMID 8505985. S2CID 4254940.
- ^ Araten DJ, Golde DW, Zhang RH, Thaler HT, Gargiulo L, Notaro R, Luzzatto L (September 2005). «A quantitative measurement of the human somatic mutation rate». Cancer Research. 65 (18): 8111–7. doi:10.1158/0008-5472.CAN-04-1198. PMID 16166284.
- ^ «‘Lifeless’ prion proteins are ‘capable of evolution’«. Health. BBC News. London. 1 January 2010. Archived from the original on 25 September 2015. Retrieved 10 October 2015.
- ^ Sullivan AD, Wigginton J, Kirschner D (August 2001). «The coreceptor mutation CCR5Delta32 influences the dynamics of HIV epidemics and is selected for by HIV». Proceedings of the National Academy of Sciences of the United States of America. 98 (18): 10214–9. Bibcode:2001PNAS…9810214S. doi:10.1073/pnas.181325198. PMC 56941. PMID 11517319.
- ^ «Mystery of the Black Death». Secrets of the Dead. Season 3. Episode 2. 30 October 2002. PBS. Archived from the original on 12 October 2015. Retrieved 10 October 2015. Episode background.
- ^ Galvani AP, Slatkin M (December 2003). «Evaluating plague and smallpox as historical selective pressures for the CCR5-Delta 32 HIV-resistance allele». Proceedings of the National Academy of Sciences of the United States of America. 100 (25): 15276–9. Bibcode:2003PNAS..10015276G. doi:10.1073/pnas.2435085100. PMC 299980. PMID 14645720.
- ^ Konotey-Ahulu F. «Frequently Asked Questions [FAQ’s]». sicklecell.md. Archived from the original on 30 April 2011. Retrieved 16 April 2010.
- ^ Hughes D, Andersson DI (September 2017). «Evolutionary Trajectories to Antibiotic Resistance». Annual Review of Microbiology. 71: 579–596. doi:10.1146/annurev-micro-090816-093813. PMID 28697667.
- ^ Ségurel L, Bon C (August 2017). «On the Evolution of Lactase Persistence in Humans». Annual Review of Genomics and Human Genetics. 18: 297–319. doi:10.1146/annurev-genom-091416-035340. PMID 28426286.
- ^ a b c Barešić, Anja; Martin, Andrew C.R. (1 August 2011). «Compensated pathogenic deviations». BioMolecular Concepts. 2 (4): 281–292. doi:10.1515/bmc.2011.025. ISSN 1868-503X. PMID 25962036. S2CID 6540447.
- ^ a b c Whitlock, Michael C.; Griswold, Cortland K.; Peters, Andrew D. (2003). «Compensating for the meltdown: The critical effective size of a population with deleterious and compensatory mutations». Annales Zoologici Fennici. 40 (2): 169–183. ISSN 0003-455X. JSTOR 23736523.
- ^ a b Lanfear, Robert; Kokko, Hanna; Eyre-Walker, Adam (1 January 2014). «Population size and the rate of evolution». Trends in Ecology & Evolution. 29 (1): 33–41. doi:10.1016/j.tree.2013.09.009. ISSN 0169-5347. PMID 24148292.
- ^ Doudna, Jennifer A. (1 November 2000). «Structural genomics of RNA». Nature Structural Biology. 7: 954–956. doi:10.1038/80729. PMID 11103998. S2CID 998448.
- ^ Cowperthwaite, Matthew C.; Bull, J. J.; Meyers, Lauren Ancel (20 October 2006). «From Bad to Good: Fitness Reversals and the Ascent of Deleterious Mutations». PLOS Computational Biology. 2 (10): e141. Bibcode:2006PLSCB…2..141C. doi:10.1371/journal.pcbi.0020141. ISSN 1553-7358. PMC 1617134. PMID 17054393.
- ^ Cowperthwaite, Matthew C.; Meyers, Lauren Ancel (1 December 2007). «How Mutational Networks Shape Evolution: Lessons from RNA Models». Annual Review of Ecology, Evolution, and Systematics. 38 (1): 203–230. doi:10.1146/annurev.ecolsys.38.091206.095507. ISSN 1543-592X.
- ^ a b Azbukina, Nadezhda; Zharikova, Anastasia; Ramensky, Vasily (1 October 2022). «Intragenic compensation through the lens of deep mutational scanning». Biophysical Reviews. 14 (5): 1161–1182. doi:10.1007/s12551-022-01005-w. ISSN 1867-2469. PMC 9636336. PMID 36345285.
- ^ a b c d DePristo, Mark A.; Weinreich, Daniel M.; Hartl, Daniel L. (September 2005). «Missense meanderings in sequence space: a biophysical view of protein evolution». Nature Reviews. Genetics. 6 (9): 678–687. doi:10.1038/nrg1672. ISSN 1471-0056. PMID 16074985. S2CID 13236893.
- ^ a b Ferrer-Costa, Carles; Orozco, Modesto; Cruz, Xavier de la (5 January 2007). «Characterization of Compensated Mutations in Terms of Structural and Physico-Chemical Properties». Journal of Molecular Biology. 365 (1): 249–256. doi:10.1016/j.jmb.2006.09.053. ISSN 0022-2836. PMID 17059831.
- ^ Lunzer, Mark; Golding, G. Brian; Dean, Antony M. (21 October 2010). «Pervasive Cryptic Epistasis in Molecular Evolution». PLOS Genetics. 6 (10): e1001162. doi:10.1371/journal.pgen.1001162. ISSN 1553-7404. PMC 2958800. PMID 20975933.
- ^ a b Corrigan, Rebecca M.; Abbott, James C.; Burhenne, Heike; Kaever, Volkhard; Gründling, Angelika (1 September 2011). «c-di-AMP Is a New Second Messenger in Staphylococcus aureus with a Role in Controlling Cell Size and Envelope Stress». PLOS Pathogens. 7 (9): e1002217. doi:10.1371/journal.ppat.1002217. ISSN 1553-7366. PMC 3164647. PMID 21909268.
- ^ a b c Comas, Iñaki; Borrell, Sonia; Roetzer, Andreas; Rose, Graham; Malla, Bijaya; Kato-Maeda, Midori; Galagan, James; Niemann, Stefan; Gagneux, Sebastien (January 2012). «Whole-genome sequencing of rifampicin-resistant Mycobacterium tuberculosis strains identifies compensatory mutations in RNA polymerase genes». Nature Genetics. 44 (1): 106–110. doi:10.1038/ng.1038. ISSN 1546-1718. PMC 3246538. PMID 22179134.
- ^ a b Gagneux, Sebastien; Long, Clara Davis; Small, Peter M.; Van, Tran; Schoolnik, Gary K.; Bohannan, Brendan J. M. (30 June 2006). «The competitive cost of antibiotic resistance in Mycobacterium tuberculosis». Science. 312 (5782): 1944–1946. doi:10.1126/science.1124410. ISSN 1095-9203. PMID 16809538. S2CID 7454895.
- ^ a b c Reynolds, M. G. (December 2000). «Compensatory evolution in rifampin-resistant Escherichia coli». Genetics. 156 (4): 1471–1481. doi:10.1093/genetics/156.4.1471. ISSN 0016-6731. PMC 1461348. PMID 11102350.
- ^ Gong, Lizhi Ian; Suchard, Marc A; Bloom, Jesse D (14 May 2013). Pascual, Mercedes (ed.). «Stability-mediated epistasis constrains the evolution of an influenza protein». eLife. 2: e00631. doi:10.7554/eLife.00631. ISSN 2050-084X. PMC 3654441. PMID 23682315.
- ^ a b Davis, Brad H.; Poon, Art F.Y.; Whitlock, Michael C. (22 May 2009). «Compensatory mutations are repeatable and clustered within proteins». Proceedings of the Royal Society B: Biological Sciences. 276 (1663): 1823–1827. doi:10.1098/rspb.2008.1846. ISSN 0962-8452. PMC 2674493. PMID 19324785.
- ^ Azbukina, Nadezhda; Zharikova, Anastasia; Ramensky, Vasily (1 October 2022). «Intragenic compensation through the lens of deep mutational scanning». Biophysical Reviews. 14 (5): 1161–1182. doi:10.1007/s12551-022-01005-w. ISSN 1867-2469. PMC 9636336. PMID 36345285.
- ^ a b Burch, Christina L; Chao, Lin (1 March 1999). «Evolution by Small Steps and Rugged Landscapes in the RNA Virus ϕ6». Genetics. 151 (3): 921–927. doi:10.1093/genetics/151.3.921. ISSN 1943-2631. PMC 1460516. PMID 10049911.
- ^ a b Rimmelzwaan, G. F.; Berkhoff, E. G. M.; Nieuwkoop, N. J.; Smith, D. J.; Fouchier, R. A. M.; Osterhaus, A. D. M. E.YR 2005 (2005). «Full restoration of viral fitness by multiple compensatory co-mutations in the nucleoprotein of influenza A virus cytotoxic T-lymphocyte escape mutants». Journal of General Virology. 86 (6): 1801–1805. doi:10.1099/vir.0.80867-0. ISSN 1465-2099. PMID 15914859.
- ^ Kimura, Motoo (1 July 1985). «The role of compensatory neutral mutations in molecular evolution». Journal of Genetics. 64 (1): 7–19. doi:10.1007/BF02923549. ISSN 0973-7731. S2CID 129866.
- ^ a b c Bowler PJ (1992) [1983]. The Eclipse of Darwinism. p. 198.
- ^ Smocovitis VB (1996). «Unifying biology: the evolutionary synthesis and evolutionary biology». Journal of the History of Biology. Princeton University Press. 25 (1): 1–65. doi:10.1007/bf01947504. ISBN 978-0-691-03343-3. LCCN 96005605. OCLC 34411399. PMID 11623198. S2CID 189833728.
- ^ Hallgrímsson B, Hall BK (2011). Variation: A Central Concept in Biology. Academic Press. p. 18.
- ^ Sewall Wright. (1984). Evolution and the Genetics of Populations: Genetics and Biometric Foundations Volume 1. University of Chicago Press. p. 10
- ^ De Vries H (1905). Species and Varieties: Their Origin by Mutation.
- ^ Bateson W (1894). Materials for the Study of Variation, Treated with Especial Regard to Discontinuity in the Origin of Species.
- ^ Punnett RC (1915). Mimicry in Butterflies. Cambridge University Press.
- ^ Mayr E (2007). What Makes Biology Unique?: Considerations on the Autonomy of a Scientific Discipline. Cambridge University Press.
- ^ a b Provine WB (2001). The Origins of Theoretical Population Genetics, with a new afterword. University of Chicago Press, Chicago. pp. 56–107.
- ^ a b Stoltzfus A, Cable K (2014). «Mendelian-mutationism: the forgotten evolutionary synthesis». Journal of the History of Biology. 47 (4): 501–46. doi:10.1007/s10739-014-9383-2. PMID 24811736. S2CID 23263558.
- ^ Hull DL (1985). «Darwinism as an historical entity: A historiographic proposal». In Kohn D (ed.). The Darwinian Heritage. Princeton University Press. pp. 773–812. ISBN 9780691024141.
- ^ Yule GU (1902). «Mendel’s Laws and their probable relations to inter-racial heredity». New Phytologist. 1 (10): 226–227. doi:10.1111/j.1469-8137.1902.tb07336.x.
- ^ Gould SJ (1982). The uses of heresey; an introduction to Richard Goldschmidt’s The Material Basis of Evolution. Yale University Press. pp. xiii–xlii. ISBN 0300028237.
- ^ Ruse M (1996). Monad to man: the Concept of Progress in Evolutionary Biology. Harvard University Press. pp. 412–413. ISBN 978-0-674-03248-4.
- ^ Stoltzfus A (2014). «In search of mutation-driven evolution». Evolution & Development. 16: 57–59. doi:10.1111/ede.12062.
- ^ Futuyma DJ (2015). Serrelli E, Gontier N (eds.). Can Modern Evolutionary Theory Explain Macroevolution? (PDF). Macroevolution. Springer. pp. 29–85. Archived from the original (PDF) on 23 February 2020. Retrieved 31 October 2017.
External links[edit]
![]()
Wikimedia Commons has media related to Mutations.
- Jones S, Woolfson A, Partridge L (6 December 2007). «Genetic Mutation». In Our Time. BBC Radio 4. Retrieved 18 October 2015.
- Liou S (5 February 2011). «All About Mutations». HOPES. Huntington’s Disease Outreach Project for Education at Stanford. Retrieved 18 October 2015.
- «Locus Specific Mutation Databases». Leiden, the Netherlands: Leiden University Medical Center. Retrieved 18 October 2015.
- «Welcome to the Mutalyzer website». Leiden, the Netherlands: Leiden University Medical Center. Retrieved 18 October 2015. – The Mutalyzer website.
A red tulip exhibiting a partially yellow petal due to a mutation in its genes
Mutation with double bloom in the Langheck Nature Reserve near Nittel, Germany
In biology, a mutation is an alteration in the nucleic acid sequence of the genome of an organism, virus, or extrachromosomal DNA.[1] Viral genomes contain either DNA or RNA. Mutations result from errors during DNA or viral replication, mitosis, or meiosis or other types of damage to DNA (such as pyrimidine dimers caused by exposure to ultraviolet radiation), which then may undergo error-prone repair (especially microhomology-mediated end joining),[2] cause an error during other forms of repair,[3][4] or cause an error during replication (translesion synthesis). Mutations may also result from insertion or deletion of segments of DNA due to mobile genetic elements.[5][6][7]
Mutations may or may not produce detectable changes in the observable characteristics (phenotype) of an organism. Mutations play a part in both normal and abnormal biological processes including: evolution, cancer, and the development of the immune system, including junctional diversity. Mutation is the ultimate source of all genetic variation, providing the raw material on which evolutionary forces such as natural selection can act.
Mutation can result in many different types of change in sequences. Mutations in genes can have no effect, alter the product of a gene, or prevent the gene from functioning properly or completely. Mutations can also occur in non-genic regions. A 2007 study on genetic variations between different species of Drosophila suggested that, if a mutation changes a protein produced by a gene, the result is likely to be harmful, with an estimated 70% of amino acid polymorphisms that have damaging effects, and the remainder being either neutral or marginally beneficial.[8] Due to the damaging effects that mutations can have on genes, organisms have mechanisms such as DNA repair to prevent or correct mutations by reverting the mutated sequence back to its original state.[5]
Overview[edit]
Mutations can involve the duplication of large sections of DNA, usually through genetic recombination.[9] These duplications are a major source of raw material for evolving new genes, with tens to hundreds of genes duplicated in animal genomes every million years.[10] Most genes belong to larger gene families of shared ancestry, detectable by their sequence homology.[11] Novel genes are produced by several methods, commonly through the duplication and mutation of an ancestral gene, or by recombining parts of different genes to form new combinations with new functions.[12][13]
Here, protein domains act as modules, each with a particular and independent function, that can be mixed together to produce genes encoding new proteins with novel properties.[14] For example, the human eye uses four genes to make structures that sense light: three for cone cell or color vision and one for rod cell or night vision; all four arose from a single ancestral gene.[15] Another advantage of duplicating a gene (or even an entire genome) is that this increases engineering redundancy; this allows one gene in the pair to acquire a new function while the other copy performs the original function.[16][17] Other types of mutation occasionally create new genes from previously noncoding DNA.[18][19]
Changes in chromosome number may involve even larger mutations, where segments of the DNA within chromosomes break and then rearrange. For example, in the Homininae, two chromosomes fused to produce human chromosome 2; this fusion did not occur in the lineage of the other apes, and they retain these separate chromosomes.[20] In evolution, the most important role of such chromosomal rearrangements may be to accelerate the divergence of a population into new species by making populations less likely to interbreed, thereby preserving genetic differences between these populations.[21]
Sequences of DNA that can move about the genome, such as transposons, make up a major fraction of the genetic material of plants and animals, and may have been important in the evolution of genomes.[22] For example, more than a million copies of the Alu sequence are present in the human genome, and these sequences have now been recruited to perform functions such as regulating gene expression.[23] Another effect of these mobile DNA sequences is that when they move within a genome, they can mutate or delete existing genes and thereby produce genetic diversity.[6]
Nonlethal mutations accumulate within the gene pool and increase the amount of genetic variation.[24] The abundance of some genetic changes within the gene pool can be reduced by natural selection, while other «more favorable» mutations may accumulate and result in adaptive changes.
For example, a butterfly may produce offspring with new mutations. The majority of these mutations will have no effect; but one might change the color of one of the butterfly’s offspring, making it harder (or easier) for predators to see. If this color change is advantageous, the chances of this butterfly’s surviving and producing its own offspring are a little better, and over time the number of butterflies with this mutation may form a larger percentage of the population.
Neutral mutations are defined as mutations whose effects do not influence the fitness of an individual. These can increase in frequency over time due to genetic drift. It is believed that the overwhelming majority of mutations have no significant effect on an organism’s fitness.[25][26] Also, DNA repair mechanisms are able to mend most changes before they become permanent mutations, and many organisms have mechanisms for eliminating otherwise-permanently mutated somatic cells.
Beneficial mutations can improve reproductive success.[27][28]
Causes[edit]
Four classes of mutations are (1) spontaneous mutations (molecular decay), (2) mutations due to error-prone replication bypass of naturally occurring DNA damage (also called error-prone translesion synthesis), (3) errors introduced during DNA repair, and (4) induced mutations caused by mutagens. Scientists may also deliberately introduce mutant sequences through DNA manipulation for the sake of scientific experimentation.
One 2017 study claimed that 66% of cancer-causing mutations are random, 29% are due to the environment (the studied population spanned 69 countries), and 5% are inherited.[29]
Humans on average pass 60 new mutations to their children but fathers pass more mutations depending on their age with every year adding two new mutations to a child.[30]
Spontaneous mutation[edit]
Spontaneous mutations occur with non-zero probability even given a healthy, uncontaminated cell. Naturally occurring oxidative DNA damage is estimated to occur 10,000 times per cell per day in humans and 100,000 times per cell per day in rats.[31] Spontaneous mutations can be characterized by the specific change:[32]
- Tautomerism – A base is changed by the repositioning of a hydrogen atom, altering the hydrogen bonding pattern of that base, resulting in incorrect base pairing during replication.[33] Theoretical results suggest that proton tunneling is an important factor in the spontaneous creation of GC tautomers.[34]
- Depurination – Loss of a purine base (A or G) to form an apurinic site (AP site).
- Deamination – Hydrolysis changes a normal base to an atypical base containing a keto group in place of the original amine group. Examples include C → U and A → HX (hypoxanthine), which can be corrected by DNA repair mechanisms; and 5MeC (5-methylcytosine) → T, which is less likely to be detected as a mutation because thymine is a normal DNA base.
- Slipped strand mispairing – Denaturation of the new strand from the template during replication, followed by renaturation in a different spot («slipping»). This can lead to insertions or deletions.
Error-prone replication bypass[edit]
There is increasing evidence that the majority of spontaneously arising mutations are due to error-prone replication (translesion synthesis) past DNA damage in the template strand. In mice, the majority of mutations are caused by translesion synthesis.[35] Likewise, in yeast, Kunz et al.[36] found that more than 60% of the spontaneous single base pair substitutions and deletions were caused by translesion synthesis.
Errors introduced during DNA repair[edit]
Although naturally occurring double-strand breaks occur at a relatively low frequency in DNA, their repair often causes mutation. Non-homologous end joining (NHEJ) is a major pathway for repairing double-strand breaks. NHEJ involves removal of a few nucleotides to allow somewhat inaccurate alignment of the two ends for rejoining followed by addition of nucleotides to fill in gaps. As a consequence, NHEJ often introduces mutations.[37]
Induced mutation[edit]
Induced mutations are alterations in the gene after it has come in contact with mutagens and environmental causes.
Induced mutations on the molecular level can be caused by:
- Chemicals
- Hydroxylamine
- Base analogs (e.g., Bromodeoxyuridine (BrdU))
- Alkylating agents (e.g., N-ethyl-N-nitrosourea (ENU). These agents can mutate both replicating and non-replicating DNA. In contrast, a base analog can mutate the DNA only when the analog is incorporated in replicating the DNA. Each of these classes of chemical mutagens has certain effects that then lead to transitions, transversions, or deletions.
- Agents that form DNA adducts (e.g., ochratoxin A)[39]
- DNA intercalating agents (e.g., ethidium bromide)
- DNA crosslinkers
- Oxidative damage
- Nitrous acid converts amine groups on A and C to diazo groups, altering their hydrogen bonding patterns, which leads to incorrect base pairing during replication.
- Radiation
- Ultraviolet light (UV) (including non-ionizing radiation). Two nucleotide bases in DNA—cytosine and thymine—are most vulnerable to radiation that can change their properties. UV light can induce adjacent pyrimidine bases in a DNA strand to become covalently joined as a pyrimidine dimer. UV radiation, in particular longer-wave UVA, can also cause oxidative damage to DNA.[40]
- Ionizing radiation. Exposure to ionizing radiation, such as gamma radiation, can result in mutation, possibly resulting in cancer or death.
Whereas in former times mutations were assumed to occur by chance, or induced by mutagens, molecular mechanisms of mutation have been discovered in bacteria and across the tree of life. As S. Rosenberg states, «These mechanisms reveal a picture of highly regulated mutagenesis, up-regulated temporally by stress responses and activated when cells/organisms are maladapted to their environments—when stressed—potentially accelerating adaptation.»[41] Since they are self-induced mutagenic mechanisms that increase the adaptation rate of organisms, they have some times been named as adaptive mutagenesis mechanisms, and include the SOS response in bacteria,[42] ectopic intrachromosomal recombination[43] and other chromosomal events such as duplications.[41]
Classification of types[edit]
By effect on structure[edit]
![]()
Five types of chromosomal mutations
![]()
Types of small-scale mutations
The sequence of a gene can be altered in a number of ways.[44] Gene mutations have varying effects on health depending on where they occur and whether they alter the function of essential proteins.
Mutations in the structure of genes can be classified into several types.
Large-scale mutations[edit]
Large-scale mutations in chromosomal structure include:
- Amplifications (or gene duplications) or repetition of a chromosomal segment or presence of extra piece of a chromosome broken piece of a chromosome may become attached to a homologous or non-homologous chromosome so that some of the genes are present in more than two doses leading to multiple copies of all chromosomal regions, increasing the dosage of the genes located within them.
- Polyploidy, duplication of entire sets of chromosomes, potentially resulting in a separate breeding population and speciation.
- Deletions of large chromosomal regions, leading to loss of the genes within those regions.
- Mutations whose effect is to juxtapose previously separate pieces of DNA, potentially bringing together separate genes to form functionally distinct fusion genes (e.g., bcr-abl).
- Large scale changes to the structure of chromosomes called chromosomal rearrangement that can lead to a decrease of fitness but also to speciation in isolated, inbred populations. These include:
- Chromosomal translocations: interchange of genetic parts from nonhomologous chromosomes.
- Chromosomal inversions: reversing the orientation of a chromosomal segment.
- Non-homologous chromosomal crossover.
- Interstitial deletions: an intra-chromosomal deletion that removes a segment of DNA from a single chromosome, thereby apposing previously distant genes. For example, cells isolated from a human astrocytoma, a type of brain tumor, were found to have a chromosomal deletion removing sequences between the Fused in Glioblastoma (FIG) gene and the receptor tyrosine kinase (ROS), producing a fusion protein (FIG-ROS). The abnormal FIG-ROS fusion protein has constitutively active kinase activity that causes oncogenic transformation (a transformation from normal cells to cancer cells).
- Loss of heterozygosity: loss of one allele, either by a deletion or a genetic recombination event, in an organism that previously had two different alleles.
Small-scale mutations[edit]
Small-scale mutations affect a gene in one or a few nucleotides. (If only a single nucleotide is affected, they are called point mutations.) Small-scale mutations include:
- Insertions add one or more extra nucleotides into the DNA. They are usually caused by transposable elements, or errors during replication of repeating elements. Insertions in the coding region of a gene may alter splicing of the mRNA (splice site mutation), or cause a shift in the reading frame (frameshift), both of which can significantly alter the gene product. Insertions can be reversed by excision of the transposable element.
- Deletions remove one or more nucleotides from the DNA. Like insertions, these mutations can alter the reading frame of the gene. In general, they are irreversible: Though exactly the same sequence might, in theory, be restored by an insertion, transposable elements able to revert a very short deletion (say 1–2 bases) in any location either are highly unlikely to exist or do not exist at all.
- Substitution mutations, often caused by chemicals or malfunction of DNA replication, exchange a single nucleotide for another.[45] These changes are classified as transitions or transversions.[46] Most common is the transition that exchanges a purine for a purine (A ↔ G) or a pyrimidine for a pyrimidine, (C ↔ T). A transition can be caused by nitrous acid, base mispairing, or mutagenic base analogs such as BrdU. Less common is a transversion, which exchanges a purine for a pyrimidine or a pyrimidine for a purine (C/T ↔ A/G). An example of a transversion is the conversion of adenine (A) into a cytosine (C). Point mutations are modifications of single base pairs of DNA or other small base pairs within a gene. A point mutation can be reversed by another point mutation, in which the nucleotide is changed back to its original state (true reversion) or by second-site reversion (a complementary mutation elsewhere that results in regained gene functionality). As discussed below, point mutations that occur within the protein coding region of a gene may be classified as synonymous or nonsynonymous substitutions, the latter of which in turn can be divided into missense or nonsense mutations.
By impact on protein sequence[edit]
![]()
Point mutations classified by impact on protein
![]()
The effect of a mutation on protein sequence depends in part on where in the genome it occurs, especially whether it is in a coding or non-coding region. Mutations in the non-coding regulatory sequences of a gene, such as promoters, enhancers, and silencers, can alter levels of gene expression, but are less likely to alter the protein sequence. Mutations within introns and in regions with no known biological function (e.g. pseudogenes, retrotransposons) are generally neutral, having no effect on phenotype – though intron mutations could alter the protein product if they affect mRNA splicing.
Mutations that occur in coding regions of the genome are more likely to alter the protein product, and can be categorized by their effect on amino acid sequence:
- A frameshift mutation is caused by insertion or deletion of a number of nucleotides that is not evenly divisible by three from a DNA sequence. Due to the triplet nature of gene expression by codons, the insertion or deletion can disrupt the reading frame, or the grouping of the codons, resulting in a completely different translation from the original.[48] The earlier in the sequence the deletion or insertion occurs, the more altered the protein produced is. (For example, the code CCU GAC UAC CUA codes for the amino acids proline, aspartic acid, tyrosine, and leucine. If the U in CCU was deleted, the resulting sequence would be CCG ACU ACC UAx, which would instead code for proline, threonine, threonine, and part of another amino acid or perhaps a stop codon (where the x stands for the following nucleotide).) By contrast, any insertion or deletion that is evenly divisible by three is termed an in-frame mutation.
- A point substitution mutation results in a change in a single nucleotide and can be either synonymous or nonsynonymous.
- A synonymous substitution replaces a codon with another codon that codes for the same amino acid, so that the produced amino acid sequence is not modified. Synonymous mutations occur due to the degenerate nature of the genetic code. If this mutation does not result in any phenotypic effects, then it is called silent, but not all synonymous substitutions are silent. (There can also be silent mutations in nucleotides outside of the coding regions, such as the introns, because the exact nucleotide sequence is not as crucial as it is in the coding regions, but these are not considered synonymous substitutions.)
- A nonsynonymous substitution replaces a codon with another codon that codes for a different amino acid, so that the produced amino acid sequence is modified. Nonsynonymous substitutions can be classified as nonsense or missense mutations:
- A missense mutation changes a nucleotide to cause substitution of a different amino acid. This in turn can render the resulting protein nonfunctional. Such mutations are responsible for diseases such as Epidermolysis bullosa, sickle-cell disease, and SOD1-mediated ALS.[49] On the other hand, if a missense mutation occurs in an amino acid codon that results in the use of a different, but chemically similar, amino acid, then sometimes little or no change is rendered in the protein. For example, a change from AAA to AGA will encode arginine, a chemically similar molecule to the intended lysine. In this latter case the mutation will have little or no effect on phenotype and therefore be neutral.
- A nonsense mutation is a point mutation in a sequence of DNA that results in a premature stop codon, or a nonsense codon in the transcribed mRNA, and possibly a truncated, and often nonfunctional protein product. This sort of mutation has been linked to different diseases, such as congenital adrenal hyperplasia. (See Stop codon.)
By effect on function[edit]
A mutation becomes an effect on function mutation when the exactitude of functions between a mutated protein and its direct interactor undergoes change. The interactors can be other proteins, molecules, nucleic acids, etc. There are many mutations that fall under the category of by effect on function, but depending on the specificity of the change the mutations listed below will occur.[50]
- Loss-of-function mutations, also called inactivating mutations, result in the gene product having less or no function (being partially or wholly inactivated). When the allele has a complete loss of function (null allele), it is often called an amorph or amorphic mutation in Muller’s morphs schema. Phenotypes associated with such mutations are most often recessive. Exceptions are when the organism is haploid, or when the reduced dosage of a normal gene product is not enough for a normal phenotype (this is called haploinsufficiency). A disease that is caused by a loss-of-function mutation is Gitelman syndrome and cystic fibrosis.[51]
- Gain-of-function mutations also called activating mutations, change the gene product such that its effect gets stronger (enhanced activation) or even is superseded by a different and abnormal function. When the new allele is created, a heterozygote containing the newly created allele as well as the original will express the new allele; genetically this defines the mutations as dominant phenotypes. Several of Muller’s morphs correspond to the gain of function, including hypermorph (increased gene expression) and neomorph (novel function). In December 2017, the U.S. government lifted a temporary ban implemented in 2014 that banned federal funding for any new «gain-of-function» experiments that enhance pathogens «such as Avian influenza, SARS, and the Middle East Respiratory Syndrome or MERS viruses. Many diseases are caused by this mutation including systemic mastocytosis and STAT3 disease.[52]
- Dominant negative mutations (also called anti-morphic mutations) have an altered gene product that acts antagonistically to the wild-type allele. These mutations usually result in an altered molecular function (often inactive) and are characterized by a dominant or semi-dominant phenotype. In humans, dominant negative mutations have been implicated in cancer (e.g., mutations in genes p53, ATM, CEBPA, and PPARgamma]). Marfan syndrome is caused by mutations in the FBN1 gene, located on chromosome 15, which encodes fibrillin-1, a glycoprotein component of the extracellular matrix. Marfan syndrome is also an example of dominant negative mutation and haploinsufficiency.
- Lethal mutations result in the instant death of the developing organism. Lethal mutations can also lead to a substantial loss in the life expectancy of the organism. An example of a disease that is caused by a dominant lethal mutation is Huntington’s disease.
- Null mutations, also known as Amorphic mutations, are a form of loss-of-function mutations that completely prohibit the gene’s function. The mutation leads to a complete loss of operation at the phenotypic level, also causing no gene product to be formed. Atopic eczema and dermatitis syndrome are common diseases caused by a null mutation of the gene that activates filaggrin.
- Suppressor mutations are a type of mutation that causes the double mutation to appear normally. In suppressor mutations the phenotypic activity of a different mutation is completely suppressed, thus causing the double mutation to look normal. There are two types of suppressor mutations, there are intragenic and extragenic suppressor mutations. Intragenic mutations occur in the gene where the first mutation occurs, while extragenic mutations occur in the gene that interacts with the product of the first mutation. A common disease that results from this type of mutation is Alzheimer’s disease.[53]
- Neomorphic mutations are a part of the gain-of-function mutations and are characterized by the control of new protein product synthesis. The newly synthesized gene normally contains a novel gene expression or molecular function. The result of the neomorphic mutation is the gene where the mutation occurs has a complete change in function.[54]
- A back mutation or reversion is a point mutation that restores the original sequence and hence the original phenotype.[55]
By effect on fitness (harmful, beneficial, neutral mutations)[edit]
In genetics, it is sometimes useful to classify mutations as either harmful or beneficial (or neutral):
- A harmful, or deleterious, mutation decreases the fitness of the organism. Many, but not all mutations in essential genes are harmful (if a mutation does not change the amino acid sequence in an essential protein, it is harmless in most cases).
- A beneficial, or advantageous mutation increases the fitness of the organism. Examples are mutations that lead to antibiotic resistance in bacteria (which are beneficial for bacteria but usually not for humans).
- A neutral mutation has no harmful or beneficial effect on the organism. Such mutations occur at a steady rate, forming the basis for the molecular clock. In the neutral theory of molecular evolution, neutral mutations provide genetic drift as the basis for most variation at the molecular level. In animals or plants, most mutations are neutral, given that the vast majority of their genomes is either non-coding or consists of repetitive sequences that have no obvious function («junk DNA»).[56]
Large-scale quantitative mutagenesis screens, in which thousands of millions of mutations are tested, invariably find that a larger fraction of mutations has harmful effects but always returns a number of beneficial mutations as well. For instance, in a screen of all gene deletions in E. coli, 80% of mutations were negative, but 20% were positive, even though many had a very small effect on growth (depending on condition).[57] Note that gene deletions involve removal of whole genes, so that point mutations almost always have a much smaller effect. In a similar screen in Streptococcus pneumoniae, but this time with transposon insertions, 76% of insertion mutants were classified as neutral, 16% had a significantly reduced fitness, but 6% were advantageous.[58]
This classification is obviously relative and somewhat artificial: a harmful mutation can quickly turn into a beneficial mutations when conditions change. Also, there is a gradient from harmful/beneficial to neutral, as many mutations may have small and mostly neglectable effects but under certain conditions will become relevant. Also, many traits are determined by hundreds of genes (or loci), so that each locus has only a minor effect. For instance, human height is determined by hundreds of genetic variants («mutations») but each of them has a very minor effect on height,[59] apart from the impact of nutrition. Height (or size) itself may be more or less beneficial as the huge range of sizes in animal or plant groups shows.
Distribution of fitness effects (DFE)[edit]
Attempts have been made to infer the distribution of fitness effects (DFE) using mutagenesis experiments and theoretical models applied to molecular sequence data. DFE, as used to determine the relative abundance of different types of mutations (i.e., strongly deleterious, nearly neutral or advantageous), is relevant to many evolutionary questions, such as the maintenance of genetic variation,[60] the rate of genomic decay,[61] the maintenance of outcrossing sexual reproduction as opposed to inbreeding[62] and the evolution of sex and genetic recombination.[63] DFE can also be tracked by tracking the skewness of the distribution of mutations with putatively severe effects as compared to the distribution of mutations with putatively mild or absent effect.[64] In summary, the DFE plays an important role in predicting evolutionary dynamics.[65][66] A variety of approaches have been used to study the DFE, including theoretical, experimental and analytical methods.
- Mutagenesis experiment: The direct method to investigate the DFE is to induce mutations and then measure the mutational fitness effects, which has already been done in viruses, bacteria, yeast, and Drosophila. For example, most studies of the DFE in viruses used site-directed mutagenesis to create point mutations and measure relative fitness of each mutant.[67][68][69][70] In Escherichia coli, one study used transposon mutagenesis to directly measure the fitness of a random insertion of a derivative of Tn10.[71] In yeast, a combined mutagenesis and deep sequencing approach has been developed to generate high-quality systematic mutant libraries and measure fitness in high throughput.[72] However, given that many mutations have effects too small to be detected[73] and that mutagenesis experiments can detect only mutations of moderately large effect; DNA sequence analysis can provide valuable information about these mutations.
The distribution of fitness effects (DFE) of mutations in vesicular stomatitis virus. In this experiment, random mutations were introduced into the virus by site-directed mutagenesis, and the fitness of each mutant was compared with the ancestral type. A fitness of zero, less than one, one, more than one, respectively, indicates that mutations are lethal, deleterious, neutral, and advantageous.[67]
-
This figure shows a simplified version of loss-of-function, switch-of-function, gain-of-function, and conservation-of-function mutations.
Molecular sequence analysis: With rapid development of DNA sequencing technology, an enormous amount of DNA sequence data is available and even more is forthcoming in the future. Various methods have been developed to infer the DFE from DNA sequence data.[74][75][76][77] By examining DNA sequence differences within and between species, we are able to infer various characteristics of the DFE for neutral, deleterious and advantageous mutations.[24] To be specific, the DNA sequence analysis approach allows us to estimate the effects of mutations with very small effects, which are hardly detectable through mutagenesis experiments.
One of the earliest theoretical studies of the distribution of fitness effects was done by Motoo Kimura, an influential theoretical population geneticist. His neutral theory of molecular evolution proposes that most novel mutations will be highly deleterious, with a small fraction being neutral.[25][78] A later proposal by Hiroshi Akashi proposed a bimodal model for the DFE, with modes centered around highly deleterious and neutral mutations.[79] Both theories agree that the vast majority of novel mutations are neutral or deleterious and that advantageous mutations are rare, which has been supported by experimental results. One example is a study done on the DFE of random mutations in vesicular stomatitis virus.[67] Out of all mutations, 39.6% were lethal, 31.2% were non-lethal deleterious, and 27.1% were neutral. Another example comes from a high throughput mutagenesis experiment with yeast.[72] In this experiment it was shown that the overall DFE is bimodal, with a cluster of neutral mutations, and a broad distribution of deleterious mutations.
Though relatively few mutations are advantageous, those that are play an important role in evolutionary changes.[80] Like neutral mutations, weakly selected advantageous mutations can be lost due to random genetic drift, but strongly selected advantageous mutations are more likely to be fixed. Knowing the DFE of advantageous mutations may lead to increased ability to predict the evolutionary dynamics. Theoretical work on the DFE for advantageous mutations has been done by John H. Gillespie[81] and H. Allen Orr.[82] They proposed that the distribution for advantageous mutations should be exponential under a wide range of conditions, which, in general, has been supported by experimental studies, at least for strongly selected advantageous mutations.[83][84][85]
In general, it is accepted that the majority of mutations are neutral or deleterious, with advantageous mutations being rare; however, the proportion of types of mutations varies between species. This indicates two important points: first, the proportion of effectively neutral mutations is likely to vary between species, resulting from dependence on effective population size; second, the average effect of deleterious mutations varies dramatically between species.[24] In addition, the DFE also differs between coding regions and noncoding regions, with the DFE of noncoding DNA containing more weakly selected mutations.[24]
By inheritance[edit]
A mutation has caused this moss rose plant to produce flowers of different colors. This is a somatic mutation that may also be passed on in the germline.
In multicellular organisms with dedicated reproductive cells, mutations can be subdivided into germline mutations, which can be passed on to descendants through their reproductive cells, and somatic mutations (also called acquired mutations),[86] which involve cells outside the dedicated reproductive group and which are not usually transmitted to descendants.
Diploid organisms (e.g., humans) contain two copies of each gene—a paternal and a maternal allele. Based on the occurrence of mutation on each chromosome, we may classify mutations into three types. A wild type or homozygous non-mutated organism is one in which neither allele is mutated.
- A heterozygous mutation is a mutation of only one allele.
- A homozygous mutation is an identical mutation of both the paternal and maternal alleles.
- Compound heterozygous mutations or a genetic compound consists of two different mutations in the paternal and maternal alleles.[87]
Germline mutation[edit]
A germline mutation in the reproductive cells of an individual gives rise to a constitutional mutation in the offspring, that is, a mutation that is present in every cell. A constitutional mutation can also occur very soon after fertilisation, or continue from a previous constitutional mutation in a parent.[88] A germline mutation can be passed down through subsequent generations of organisms.
The distinction between germline and somatic mutations is important in animals that have a dedicated germline to produce reproductive cells. However, it is of little value in understanding the effects of mutations in plants, which lack a dedicated germline. The distinction is also blurred in those animals that reproduce asexually through mechanisms such as budding, because the cells that give rise to the daughter organisms also give rise to that organism’s germline.
A new germline mutation not inherited from either parent is called a de novo mutation.
Somatic mutation[edit]
GENE MUTATIONS:
Gene mutations include either the replacement of one of the nucleotides with the nucleotide by the other nucleotide or may be by the addition or the deletion of the nucleotide.[89] This would be explained as the sudden change or the alteration in nucleotide sequence of the DNA molecule, which would affect one pair of nucleotide or the bigger art of the gene on chromosome.[90] These gene mutations can be further classified as:
1. Point mutations: This results when there is difference in only one base pair of nucleotide which can also be called as base pair substitution and this is also one of the common type among the gene mutations. Point mutations can be again divided into three types of mutations namely Silent mutations, Nonsense mutations, Missense mutations.
a) Silent Mutations:
This occurs when there is a change in codon for one amino acid molecule is swapped or is into the other codon of the same amino acid molecule and is also referred as “synonymous mutations”
b) Missense Mutations:
This occurs when the codon of one amino acid is interchanged with the codon of another amino acid and can also be referred as non-synonymous mutations.
c) Nonsense Mutations:
This occurs when the codon of the amino acid changes to the stop codon.
2. Frameshift Mutations:
This kind of mutation results when there is addition or deletion of DNA base molecules changes the reading frame of the gene. This mutations would be insertions or deletions.[90]
a) Insertion:
This type of mutation differs the DNA base number in the gene by adding the part of the DNA.
b) Deletion:
This type of mutation occurs when there is a difference in the number of DNA bases by eliminating a piece of DNA.
3. Base substitution Mutations:
This type of mutations occur when there is replacement of one base pair by the other base pair. This mutations are further classified as Transition mutation and transversion mutation,
a) Transition mutation: This occurs when the base of one chemical is replaced by the other base of the same chemical molecule (4). It mainly happens when there is the transposing of the purine molecules i.e., A is transposed by G or by the transposing of pyrimidine molecules i.e., C by T in the DNA molecule.
b) Tranvsersion Mutation: This occurs when there is an opposite replacement of a category base chemical by another base of the other category . This is mainly due to the incorrect replacement of the DNA bases i.e., when a pyrimidine is replaced with purine molecule.
A change in the genetic structure that is not inherited from a parent, and also not passed to offspring, is called a somatic mutation.[86] Somatic mutations are not inherited by an organism’s offspring because they do not affect the germline. However, they are passed down to all the progeny of a mutated cell within the same organism during mitosis. A major section of an organism therefore might carry the same mutation. These types of mutations are usually prompted by environmental causes, such as ultraviolet radiation or any exposure to certain harmful chemicals, and can cause diseases including cancer.[91]
With plants, some somatic mutations can be propagated without the need for seed production, for example, by grafting and stem cuttings. These type of mutation have led to new types of fruits, such as the «Delicious» apple and the «Washington» navel orange.[92]
Human and mouse somatic cells have a mutation rate more than ten times higher than the germline mutation rate for both species; mice have a higher rate of both somatic and germline mutations per cell division than humans. The disparity in mutation rate between the germline and somatic tissues likely reflects the greater importance of genome maintenance in the germline than in the soma.[93]
Special classes[edit]
- Conditional mutation is a mutation that has wild-type (or less severe) phenotype under certain «permissive» environmental conditions and a mutant phenotype under certain «restrictive» conditions. For example, a temperature-sensitive mutation can cause cell death at high temperature (restrictive condition), but might have no deleterious consequences at a lower temperature (permissive condition).[94] These mutations are non-autonomous, as their manifestation depends upon presence of certain conditions, as opposed to other mutations which appear autonomously.[95] The permissive conditions may be temperature,[96] certain chemicals,[97] light[97] or mutations in other parts of the genome.[95] In vivo mechanisms like transcriptional switches can create conditional mutations. For instance, association of Steroid Binding Domain can create a transcriptional switch that can change the expression of a gene based on the presence of a steroid ligand.[98] Conditional mutations have applications in research as they allow control over gene expression. This is especially useful studying diseases in adults by allowing expression after a certain period of growth, thus eliminating the deleterious effect of gene expression seen during stages of development in model organisms.[97] DNA Recombinase systems like Cre-Lox recombination used in association with promoters that are activated under certain conditions can generate conditional mutations. Dual Recombinase technology can be used to induce multiple conditional mutations to study the diseases which manifest as a result of simultaneous mutations in multiple genes.[97] Certain inteins have been identified which splice only at certain permissive temperatures, leading to improper protein synthesis and thus, loss-of-function mutations at other temperatures.[99] Conditional mutations may also be used in genetic studies associated with ageing, as the expression can be changed after a certain time period in the organism’s lifespan.[96]
- Replication timing quantitative trait loci affects DNA replication.
Nomenclature[edit]
In order to categorize a mutation as such, the «normal» sequence must be obtained from the DNA of a «normal» or «healthy» organism (as opposed to a «mutant» or «sick» one), it should be identified and reported; ideally, it should be made publicly available for a straightforward nucleotide-by-nucleotide comparison, and agreed upon by the scientific community or by a group of expert geneticists and biologists, who have the responsibility of establishing the standard or so-called «consensus» sequence. This step requires a tremendous scientific effort. Once the consensus sequence is known, the mutations in a genome can be pinpointed, described, and classified. The committee of the Human Genome Variation Society (HGVS) has developed the standard human sequence variant nomenclature,[100] which should be used by researchers and DNA diagnostic centers to generate unambiguous mutation descriptions. In principle, this nomenclature can also be used to describe mutations in other organisms. The nomenclature specifies the type of mutation and base or amino acid changes.
- Nucleotide substitution (e.g., 76A>T) – The number is the position of the nucleotide from the 5′ end; the first letter represents the wild-type nucleotide, and the second letter represents the nucleotide that replaced the wild type. In the given example, the adenine at the 76th position was replaced by a thymine.
- If it becomes necessary to differentiate between mutations in genomic DNA, mitochondrial DNA, and RNA, a simple convention is used. For example, if the 100th base of a nucleotide sequence mutated from G to C, then it would be written as g.100G>C if the mutation occurred in genomic DNA, m.100G>C if the mutation occurred in mitochondrial DNA, or r.100g>c if the mutation occurred in RNA. Note that, for mutations in RNA, the nucleotide code is written in lower case.
- Amino acid substitution (e.g., D111E) – The first letter is the one letter code of the wild-type amino acid, the number is the position of the amino acid from the N-terminus, and the second letter is the one letter code of the amino acid present in the mutation. Nonsense mutations are represented with an X for the second amino acid (e.g. D111X).
- Amino acid deletion (e.g., ΔF508) – The Greek letter Δ (delta) indicates a deletion. The letter refers to the amino acid present in the wild type and the number is the position from the N terminus of the amino acid were it to be present as in the wild type.
Mutation rates[edit]
Mutation rates vary substantially across species, and the evolutionary forces that generally determine mutation are the subject of ongoing investigation.
In humans, the mutation rate is about 50-90 de novo mutations per genome per generation, that is, each human accumulates about 50-90 novel mutations that were not present in his or her parents. This number has been established by sequencing thousands of human trios, that is, two parents and at least one child.[101]
The genomes of RNA viruses are based on RNA rather than DNA. The RNA viral genome can be double-stranded (as in DNA) or single-stranded. In some of these viruses (such as the single-stranded human immunodeficiency virus), replication occurs quickly, and there are no mechanisms to check the genome for accuracy. This error-prone process often results in mutations.
Randomness of mutations[edit]
There is a widespread assumption that mutations are (entirely) «random» with respect to their consequences (in terms of probability). This was shown to be wrong as mutation frequency can vary across regions of the genome, with such DNA repair- and mutation-biases being associated with various factors. For instance, biologically important regions were found to be protected from mutations and mutations beneficial to the studied plant were found to be more likely – i.e. mutation is «non-random in a way that benefits the plant».[102][103]
Disease causation[edit]
Changes in DNA caused by mutation in a coding region of DNA can cause errors in protein sequence that may result in partially or completely non-functional proteins. Each cell, in order to function correctly, depends on thousands of proteins to function in the right places at the right times. When a mutation alters a protein that plays a critical role in the body, a medical condition can result. One study on the comparison of genes between different species of Drosophila suggests that if a mutation does change a protein, the mutation will most likely be harmful, with an estimated 70 percent of amino acid polymorphisms having damaging effects, and the remainder being either neutral or weakly beneficial.[8] Some mutations alter a gene’s DNA base sequence but do not change the protein made by the gene. Studies have shown that only 7% of point mutations in noncoding DNA of yeast are deleterious and 12% in coding DNA are deleterious. The rest of the mutations are either neutral or slightly beneficial.[104]
Inherited disorders[edit]
If a mutation is present in a germ cell, it can give rise to offspring that carries the mutation in all of its cells. This is the case in hereditary diseases. In particular, if there is a mutation in a DNA repair gene within a germ cell, humans carrying such germline mutations may have an increased risk of cancer. A list of 34 such germline mutations is given in the article DNA repair-deficiency disorder. An example of one is albinism, a mutation that occurs in the OCA1 or OCA2 gene. Individuals with this disorder are more prone to many types of cancers, other disorders and have impaired vision.
DNA damage can cause an error when the DNA is replicated, and this error of replication can cause a gene mutation that, in turn, could cause a genetic disorder. DNA damages are repaired by the DNA repair system of the cell. Each cell has a number of pathways through which enzymes recognize and repair damages in DNA. Because DNA can be damaged in many ways, the process of DNA repair is an important way in which the body protects itself from disease. Once DNA damage has given rise to a mutation, the mutation cannot be repaired.
Role in carcinogenesis[edit]
On the other hand, a mutation may occur in a somatic cell of an organism. Such mutations will be present in all descendants of this cell within the same organism. The accumulation of certain mutations over generations of somatic cells is part of cause of malignant transformation, from normal cell to cancer cell.[105]
Cells with heterozygous loss-of-function mutations (one good copy of gene and one mutated copy) may function normally with the unmutated copy until the good copy has been spontaneously somatically mutated. This kind of mutation happens often in living organisms, but it is difficult to measure the rate. Measuring this rate is important in predicting the rate at which people may develop cancer.[106]
Point mutations may arise from spontaneous mutations that occur during DNA replication. The rate of mutation may be increased by mutagens. Mutagens can be physical, such as radiation from UV rays, X-rays or extreme heat, or chemical (molecules that misplace base pairs or disrupt the helical shape of DNA). Mutagens associated with cancers are often studied to learn about cancer and its prevention.
Prion mutations[edit]
Prions are proteins and do not contain genetic material. However, prion replication has been shown to be subject to mutation and natural selection just like other forms of replication.[107] The human gene PRNP codes for the major prion protein, PrP, and is subject to mutations that can give rise to disease-causing prions.
Beneficial mutations[edit]
Although mutations that cause changes in protein sequences can be harmful to an organism, on occasions the effect may be positive in a given environment. In this case, the mutation may enable the mutant organism to withstand particular environmental stresses better than wild-type organisms, or reproduce more quickly. In these cases a mutation will tend to become more common in a population through natural selection. Examples include the following:
HIV resistance: a specific 32 base pair deletion in human CCR5 (CCR5-Δ32) confers HIV resistance to homozygotes and delays AIDS onset in heterozygotes.[108] One possible explanation of the etiology of the relatively high frequency of CCR5-Δ32 in the European population is that it conferred resistance to the bubonic plague in mid-14th century Europe. People with this mutation were more likely to survive infection; thus its frequency in the population increased.[109] This theory could explain why this mutation is not found in Southern Africa, which remained untouched by bubonic plague. A newer theory suggests that the selective pressure on the CCR5 Delta 32 mutation was caused by smallpox instead of the bubonic plague.[110]
Malaria resistance: An example of a harmful mutation is sickle-cell disease, a blood disorder in which the body produces an abnormal type of the oxygen-carrying substance hemoglobin in the red blood cells. One-third of all indigenous inhabitants of Sub-Saharan Africa carry the allele, because, in areas where malaria is common, there is a survival value in carrying only a single sickle-cell allele (sickle cell trait).[111] Those with only one of the two alleles of the sickle-cell disease are more resistant to malaria, since the infestation of the malaria Plasmodium is halted by the sickling of the cells that it infests.
Antibiotic resistance: Practically all bacteria develop antibiotic resistance when exposed to antibiotics. In fact, bacterial populations already have such mutations that get selected under antibiotic selection.[112] Obviously, such mutations are only beneficial for the bacteria but not for those infected.
Lactase persistence. A mutation allowed humans to express the enzyme lactase after they are naturally weaned from breast milk, allowing adults to digest lactose, which is likely one of the most beneficial mutations in recent human evolution.[113]
Compensated pathogenic deviations[edit]
Compensated pathogenic deviations refer to amino acid residues in a protein sequence that are pathogenic in one species but are wild type residues in the functionally equivalent protein in another species. Although the amino acid residue is pathogenic in the first species, it is not so in the second species because its pathogenicity is compensated by one or more amino acid substitutions in the second species. The compensatory mutation can occur in the same protein or in another protein with which it interacts.[114]
It is critical to understand the effects of compensatory mutations in the context of fixed deleterious mutations due to the population fitness decreasing because of fixation.[115] Effective population size refers to a population that is reproducing.[116] An increase in this population size has been correlated with a decreased rate of genetic diversity.[116] The position of a population relative to the critical effect population size is essential to determine the effect deleterious alleles will have on fitness.[115] If the population is below the critical effective size fitness will decrease drastically, however if the population is above the critical effect size, fitness can increase regardless of deleterious mutations due to compensatory alleles.[115]
Compensatory mutations in RNA[edit]
As the function of a RNA molecule is dependent on its structure,[117] the structure of RNA molecules is evolutionarily conserved. Therefore, any mutation that alters the stable structure of RNA molecules must be compensated by other compensatory mutations. In the context of RNA, the sequence of the RNA can be considered as ‘ genotype’ and the structure of the RNA can be considered as its ‘phenotype’. Since RNAs have relatively simpler composition than proteins, the structure of RNA molecules can be computationally predicted with high degree of accuracy. Because of this convenience, compensatory mutations have been studied in computational simulations using RNA folding algorithms.[118][119]
Evolutionary mechanism of compensation[edit]
Compensatory mutations can be explained by the genetic phenomenon epistasis whereby the phenotypic effect of one mutation is dependent upon mutation(s) at other loci. While epistasis was originally conceived in the context of interaction between different genes, intragenic epistasis has also been studied recently.[120] Existence of compensated pathogenic deviations can be explained by ‘sign epistasis’, in which the effects of a deleterious mutation can be compensated by the presence of a epistatic mutation in another loci. For a given protein, a deleterious mutation (D) and a compensatory mutation (C) can be considered, where C can be in the same protein as D or in a different interacting protein depending on the context. The fitness effect of C itself could be neutral or somewhat deleterious such that it can still exist in the population, and the effect of D is deleterious to the extent that it cannot exist in the population. However, when C and D co-occur together, the combined fitness effect becomes neutral or positive.[114] Thus, compensatory mutations can bring novelty to proteins by forging new pathways of protein evolution : it allows individuals to travel from one fitness peak to another through the valleys of lower fitness.[120]
DePristo et al. 2005 outlined two models to explain the dynamics of compensatory pathogenic deviations (CPD).[121] In the first hypothesis P is a pathogenic amino acid mutation that and C is a neutral compensatory mutation.[121] Under these conditions, if the pathogenic mutation arises after a compensatory mutation, then P can become fixed in the population.[121] The second model of CPDs states that P and C are both deleterious mutations resulting in fitness valleys when mutations occur simultaneously.[121] Using publicly available, Ferrer-Costa et al. 2007 obtained compensatory mutations and human pathogenic mutation datasets that were characterized to determine what causes CPDs.[122] Results indicate that the structural constraints and the location in protein structure determine whether compensated mutations will occur.[122]
Experimental evidence of compensatory mutations[edit]
Experiment in bacteria[edit]
Lunzer et al.[123] tested the outcome of swapping divergent amino acids between two orthologous proteins of isopropymalate dehydrogenase (IMDH). They substituted 168 amino acids in Escherichia coli IMDH that are wild type residues in IMDH Pseudomonas aeruginosa. They found that over one third of these substitutions compromised IMDH enzymatic activity in the Escherichia coli genetic background. This demonstrated that identical amino acid states can result in different phenotypic states depending on the genetic background. Corrigan et al. 2011 demonstrated how staphylococcus aureus was able to grow normally without the presence of lipoteichoic acid due to compensatory mutations.[124] Whole genome sequencing results revealed that when Cyclic-di-AMP phosphodiesterase (GdpP) was disrupted in this bacterium, it compensated for the disappearance of the cell wall polymer, resulting in normal cell growth.[124]
Research has shown that bacteria can gain drug resistance through compensatory mutations that do not impede or having little effect on fitness.[125] Previous research from Gagneux et al. 2006 has found that laboratory grown M. tuberculosis strains with rifampicin resistance have reduced fitness, however drug resistant clinical strains of this pathogenic bacteria do not have reduced fitness.[126] Comas et al. 2012 used whole genome comparisons between clinical strains and lab derived mutants to determine the role and contribution of compensatory mutations in drug resistance to rifampicin.[125] Genome analysis reveal rifampicin resistant strains have a mutation in rpoA and rpoC.[125] A similar study investigated the bacterial fitness associated with compensatory mutations in rifampin resistant Escherichia coli.[127] Results obtained from this study demonstrate that drug resistance is linked to bacterial fitness as higher fitness costs are linked to greater transcription errors.[127]
Experiment in virus[edit]
Gong et al.[128] collected obtained genotype data of influenza nucleoprotein from different timelines and temporally ordered them according to their time of origin. Then they isolated 39 amino acid substitutions that occurred in different timelines and substituted them in a genetic background that approximated the ancestral genotype. They found that 3 of the 39 substitutions significantly reduced the fitness of the ancestral background. Compensatory mutations are new mutations that arise and have a positive or neutral impact on a populations fitness.[129] Previous research has shown that populations have can compensate detrimental mutations.[114][129][130] Burch and Chao tested Fisher’s geometric model of adaptive evolution by testing whether bacteriophage φ6 evolves by small steps.[131] Their results showed that bacteriophage φ6 fitness declined rapidly and recovered in small steps .[131] Viral nucleoproteins have been shown to avoid cytotoxic T lymphocytes (CTLs) through arginine-to glycine substitutions.[132] This substitution mutations impacts the fitness of viral nucleoproteins, however compensatory co-mutations impede fitness declines and aid the virus to avoid recognition from CTLs.[132] Mutations can have three different effects; mutations can have deleterious effects, some increase fitness through compensatory mutations, and lastly mutations can be counterbalancing resulting in compensatory neutral mutations.[133][127][126]
History[edit]
Mutationism is one of several alternatives to Darwinian evolution that have existed both before and after the publication of Charles Darwin’s 1859 book, On the Origin of Species. In the theory, mutation was the source of novelty, creating new forms and new species, potentially instantaneously,[134] in a sudden jump.[135] This was envisaged as driving evolution, which was limited by the supply of mutations.
Before Darwin, biologists commonly believed in saltationism, the possibility of large evolutionary jumps, including immediate speciation. For example, in 1822 Étienne Geoffroy Saint-Hilaire argued that species could be formed by sudden transformations, or what would later be called macromutation.[136] Darwin opposed saltation, insisting on gradualism in evolution as in geology. In 1864, Albert von Kölliker revived Geoffroy’s theory.[137] In 1901 the geneticist Hugo de Vries gave the name «mutation» to seemingly new forms that suddenly arose in his experiments on the evening primrose Oenothera lamarckiana, and in the first decade of the 20th century, mutationism, or as de Vries named it mutationstheorie,[134][138] became a rival to Darwinism supported for a while by geneticists including William Bateson,[139] Thomas Hunt Morgan, and Reginald Punnett.[134][140]
Understanding of mutationism is clouded by the mid-20th century portrayal of the early mutationists by supporters of the modern synthesis as opponents of Darwinian evolution and rivals of the biometrics school who argued that selection operated on continuous variation. In this portrayal, mutationism was defeated by a synthesis of genetics and natural selection that supposedly started later, around 1918, with work by the mathematician Ronald Fisher.[141][142][143][144] However, the alignment of Mendelian genetics and natural selection began as early as 1902 with a paper by Udny Yule,[145] and built up with theoretical and experimental work in Europe and America. Despite the controversy, the early mutationists had by 1918 already accepted natural selection and explained continuous variation as the result of multiple genes acting on the same characteristic, such as height.[142][143]
Mutationism, along with other alternatives to Darwinism like Lamarckism and orthogenesis, was discarded by most biologists as they came to see that Mendelian genetics and natural selection could readily work together; mutation took its place as a source of the genetic variation essential for natural selection to work on. However, mutationism did not entirely vanish. In 1940, Richard Goldschmidt again argued for single-step speciation by macromutation, describing the organisms thus produced as «hopeful monsters», earning widespread ridicule.[146][147] In 1987, Masatoshi Nei argued controversially that evolution was often mutation-limited.[148] Modern biologists such as Douglas J. Futuyma conclude that essentially all claims of evolution driven by large mutations can be explained by Darwinian evolution.[149]
See also[edit]
- Aneuploidy
- Antioxidant
- Budgerigar colour genetics
- DbDNV (2010)
- Deletion (genetics)
- Ecogenetics
- Embryology
- Homeobox
- Human somatic variation
- Polyploidy
- Robertsonian translocation
- Signature-tagged mutagenesis
- Somatic hypermutation
- TILLING (molecular biology)
- Trinucleotide repeat expansion
References[edit]
- ^ «mutation | Learn Science at Scitable». Nature. Nature Education. Retrieved 24 September 2018.
- ^ Sfeir A, Symington LS (November 2015). «Microhomology-Mediated End Joining: A Back-up Survival Mechanism or Dedicated Pathway?». Trends in Biochemical Sciences. 40 (11): 701–714. doi:10.1016/j.tibs.2015.08.006. PMC 4638128. PMID 26439531.
- ^ Chen J, Miller BF, Furano AV (April 2014). «Repair of naturally occurring mismatches can induce mutations in flanking DNA». eLife. 3: e02001. doi:10.7554/elife.02001. PMC 3999860. PMID 24843013.
- ^ Rodgers K, McVey M (January 2016). «Error-Prone Repair of DNA Double-Strand Breaks». Journal of Cellular Physiology. 231 (1): 15–24. doi:10.1002/jcp.25053. PMC 4586358. PMID 26033759.
- ^ a b Bertram JS (December 2000). «The molecular biology of cancer». Molecular Aspects of Medicine. 21 (6): 167–223. doi:10.1016/S0098-2997(00)00007-8. PMID 11173079. S2CID 24155688.
- ^ a b Aminetzach YT, Macpherson JM, Petrov DA (July 2005). «Pesticide resistance via transposition-mediated adaptive gene truncation in Drosophila». Science. 309 (5735): 764–7. Bibcode:2005Sci…309..764A. doi:10.1126/science.1112699. PMID 16051794. S2CID 11640993.
- ^ Burrus V, Waldor MK (June 2004). «Shaping bacterial genomes with integrative and conjugative elements». Research in Microbiology. 155 (5): 376–86. doi:10.1016/j.resmic.2004.01.012. PMID 15207870.
- ^ a b Sawyer SA, Parsch J, Zhang Z, Hartl DL (April 2007). «Prevalence of positive selection among nearly neutral amino acid replacements in Drosophila». Proceedings of the National Academy of Sciences of the United States of America. 104 (16): 6504–10. Bibcode:2007PNAS..104.6504S. doi:10.1073/pnas.0701572104. PMC 1871816. PMID 17409186.
- ^ Hastings PJ, Lupski JR, Rosenberg SM, Ira G (August 2009). «Mechanisms of change in gene copy number». Nature Reviews. Genetics. 10 (8): 551–64. doi:10.1038/nrg2593. PMC 2864001. PMID 19597530.
- ^ Carroll SB, Grenier JK, Weatherbee SD (2005). From DNA to Diversity: Molecular Genetics and the Evolution of Animal Design (2nd ed.). Malden, MA: Blackwell Publishing. ISBN 978-1-4051-1950-4. LCCN 2003027991. OCLC 53972564.
- ^ Harrison PM, Gerstein M (May 2002). «Studying genomes through the aeons: protein families, pseudogenes and proteome evolution». Journal of Molecular Biology. 318 (5): 1155–74. doi:10.1016/S0022-2836(02)00109-2. PMID 12083509.
- ^ Orengo CA, Thornton JM (July 2005). «Protein families and their evolution-a structural perspective». Annual Review of Biochemistry. 74: 867–900. doi:10.1146/annurev.biochem.74.082803.133029. PMID 15954844.
- ^ Long M, Betrán E, Thornton K, Wang W (November 2003). «The origin of new genes: glimpses from the young and old». Nature Reviews. Genetics. 4 (11): 865–75. doi:10.1038/nrg1204. PMID 14634634. S2CID 33999892.
- ^ Wang M, Caetano-Anollés G (January 2009). «The evolutionary mechanics of domain organization in proteomes and the rise of modularity in the protein world». Structure. 17 (1): 66–78. doi:10.1016/j.str.2008.11.008. PMID 19141283.
- ^ Bowmaker JK (May 1998). «Evolution of colour vision in vertebrates». Eye. 12 (Pt 3b): 541–7. doi:10.1038/eye.1998.143. PMID 9775215. S2CID 12851209.
- ^ Gregory TR, Hebert PD (April 1999). «The modulation of DNA content: proximate causes and ultimate consequences». Genome Research. 9 (4): 317–24. doi:10.1101/gr.9.4.317. PMID 10207154. S2CID 16791399.
- ^ Hurles M (July 2004). «Gene duplication: the genomic trade in spare parts». PLOS Biology. 2 (7): E206. doi:10.1371/journal.pbio.0020206. PMC 449868. PMID 15252449.
- ^ Liu N, Okamura K, Tyler DM, Phillips MD, Chung WJ, Lai EC (October 2008). «The evolution and functional diversification of animal microRNA genes». Cell Research. 18 (10): 985–96. doi:10.1038/cr.2008.278. PMC 2712117. PMID 18711447.
- ^ Siepel A (October 2009). «Darwinian alchemy: Human genes from noncoding DNA». Genome Research. 19 (10): 1693–5. doi:10.1101/gr.098376.109. PMC 2765273. PMID 19797681.
- ^ Zhang J, Wang X, Podlaha O (May 2004). «Testing the chromosomal speciation hypothesis for humans and chimpanzees». Genome Research. 14 (5): 845–51. doi:10.1101/gr.1891104. PMC 479111. PMID 15123584.
- ^ Ayala FJ, Coluzzi M (May 2005). «Chromosome speciation: humans, Drosophila, and mosquitoes». Proceedings of the National Academy of Sciences of the United States of America. 102 (Suppl 1): 6535–42. Bibcode:2005PNAS..102.6535A. doi:10.1073/pnas.0501847102. PMC 1131864. PMID 15851677.
- ^ Hurst GD, Werren JH (August 2001). «The role of selfish genetic elements in eukaryotic evolution». Nature Reviews Genetics. 2 (8): 597–606. doi:10.1038/35084545. PMID 11483984. S2CID 2715605.
- ^ Häsler J, Strub K (November 2006). «Alu elements as regulators of gene expression». Nucleic Acids Research. 34 (19): 5491–7. doi:10.1093/nar/gkl706. PMC 1636486. PMID 17020921.
- ^ a b c d Eyre-Walker A, Keightley PD (August 2007). «The distribution of fitness effects of new mutations» (PDF). Nature Reviews Genetics. 8 (8): 610–8. doi:10.1038/nrg2146. PMID 17637733. S2CID 10868777. Archived from the original (PDF) on 4 March 2016. Retrieved 6 September 2010.
- ^ a b Kimura M (1983). The Neutral Theory of Molecular Evolution. Cambridge, UK; New York: Cambridge University Press. ISBN 978-0-521-23109-1. LCCN 82022225. OCLC 9081989.
- ^ Bohidar HB (January 2015). Fundamentals of Polymer Physics and Molecular Biophysics. Cambridge University Press. ISBN 978-1-316-09302-3.
- ^ Dover GA, Darwin C (2000). Dear Mr. Darwin: Letters on the Evolution of Life and Human Nature. University of California Press. ISBN 9780520227903.
- ^ Tibayrenc M (12 January 2017). Genetics and Evolution of Infectious Diseases. Elsevier. ISBN 9780128001530.
- ^ «Cancer Is Partly Caused By Bad Luck, Study Finds». NPR.org. Archived from the original on 13 July 2017.
- ^ Jha A (22 August 2012). «Older fathers pass on more genetic mutations, study shows». The Guardian.
- ^ Ames BN, Shigenaga MK, Hagen TM (September 1993). «Oxidants, antioxidants, and the degenerative diseases of aging». Proceedings of the National Academy of Sciences of the United States of America. 90 (17): 7915–22. Bibcode:1993PNAS…90.7915A. doi:10.1073/pnas.90.17.7915. PMC 47258. PMID 8367443.
- ^ Montelone BA (1998). «Mutation, Mutagens, and DNA Repair». www-personal.ksu.edu. Archived from the original on 26 September 2015. Retrieved 2 October 2015.
- ^ Slocombe L, Al-Khalili JS, Sacchi M (February 2021). «Quantum and classical effects in DNA point mutations: Watson-Crick tautomerism in AT and GC base pairs». Physical Chemistry Chemical Physics. 23 (7): 4141–4150. Bibcode:2021PCCP…23.4141S. doi:10.1039/D0CP05781A. ISSN 1463-9076. PMID 33533770. S2CID 231788542.
- ^ Slocombe L, Sacchi M, Al-Khalili J (5 May 2022). «An open quantum systems approach to proton tunnelling in DNA». Communications Physics. 5 (1): 109. arXiv:2110.00113. Bibcode:2022CmPhy…5..109S. doi:10.1038/s42005-022-00881-8. ISSN 2399-3650. S2CID 238253421.
- ^ Stuart GR, Oda Y, de Boer JG, Glickman BW (March 2000). «Mutation frequency and specificity with age in liver, bladder and brain of lacI transgenic mice». Genetics. 154 (3): 1291–300. doi:10.1093/genetics/154.3.1291. PMC 1460990. PMID 10757770.
- ^ Kunz BA, Ramachandran K, Vonarx EJ (April 1998). «DNA sequence analysis of spontaneous mutagenesis in Saccharomyces cerevisiae». Genetics. 148 (4): 1491–505. doi:10.1093/genetics/148.4.1491. PMC 1460101. PMID 9560369.
- ^ Lieber MR (July 2010). «The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway». Annual Review of Biochemistry. 79: 181–211. doi:10.1146/annurev.biochem.052308.093131. PMC 3079308. PMID 20192759.
- ^ Created from PDB 1JDG Archived 31 December 2015 at the Wayback Machine
- ^ Pfohl-Leszkowicz A, Manderville RA (January 2007). «Ochratoxin A: An overview on toxicity and carcinogenicity in animals and humans». Molecular Nutrition & Food Research. 51 (1): 61–99. doi:10.1002/mnfr.200600137. PMID 17195275.
- ^ Kozmin S, Slezak G, Reynaud-Angelin A, Elie C, de Rycke Y, Boiteux S, Sage E (September 2005). «UVA radiation is highly mutagenic in cells that are unable to repair 7,8-dihydro-8-oxoguanine in Saccharomyces cerevisiae». Proceedings of the National Academy of Sciences of the United States of America. 102 (38): 13538–43. Bibcode:2005PNAS..10213538K. doi:10.1073/pnas.0504497102. PMC 1224634. PMID 16157879.
- ^ a b Fitzgerald DM, Rosenberg SM (April 2019). «What is mutation? A chapter in the series: How microbes «jeopardize» the modern synthesis». PLOS Genetics. 15 (4): e1007995. doi:10.1371/journal.pgen.1007995. PMC 6443146. PMID 30933985.
- ^ Galhardo RS, Hastings PJ, Rosenberg SM (1 January 2007). «Mutation as a stress response and the regulation of evolvability». Critical Reviews in Biochemistry and Molecular Biology. 42 (5): 399–435. doi:10.1080/10409230701648502. PMC 3319127. PMID 17917874.
- ^ Quinto-Alemany D, Canerina-Amaro A, Hernández-Abad LG, Machín F, Romesberg FE, Gil-Lamaignere C (31 July 2012). Sturtevant J (ed.). «Yeasts acquire resistance secondary to antifungal drug treatment by adaptive mutagenesis». PLOS ONE. 7 (7): e42279. Bibcode:2012PLoSO…742279Q. doi:10.1371/journal.pone.0042279. PMC 3409178. PMID 22860105.
- ^ Rahman N. «The clinical impact of DNA sequence changes». Transforming Genetic Medicine Initiative. Archived from the original on 4 August 2017. Retrieved 27 June 2017.
- ^ Freese E (April 1959). «The Difference Between Spontaneous and Base-Analogue Induced Mutations of Phage T4». Proceedings of the National Academy of Sciences of the United States of America. 45 (4): 622–33. Bibcode:1959PNAS…45..622F. doi:10.1073/pnas.45.4.622. PMC 222607. PMID 16590424.
- ^ Freese E (June 1959). «The specific mutagenic effect of base analogues on Phage T4». Journal of Molecular Biology. 1 (2): 87–105. doi:10.1016/S0022-2836(59)80038-3.
- ^ References for the image are found in Wikimedia Commons page at: Commons:File:Notable mutations.svg#References.
- ^ Hogan CM (12 October 2010). «Mutation». In Monosson E (ed.). Encyclopedia of Earth. Washington, D.C.: Environmental Information Coalition, National Council for Science and the Environment. OCLC 72808636. Archived from the original on 14 November 2015. Retrieved 8 October 2015.
- ^ Boillée S, Vande Velde C, Cleveland DW (October 2006). «ALS: a disease of motor neurons and their nonneuronal neighbors». Neuron. 52 (1): 39–59. CiteSeerX 10.1.1.325.7514. doi:10.1016/j.neuron.2006.09.018. PMID 17015226. S2CID 12968143.
- ^ Reva B, Antipin Y, Sander C (September 2011). «Predicting the functional impact of protein mutations: application to cancer genomics». Nucleic Acids Research. 39 (17): e118. doi:10.1093/nar/gkr407. PMC 3177186. PMID 21727090.
- ^ Housden BE, Muhar M, Gemberling M, Gersbach CA, Stainier DY, Seydoux G, et al. (January 2017). «Loss-of-function genetic tools for animal models: cross-species and cross-platform differences». Nature Reviews. Genetics. 18 (1): 24–40. doi:10.1038/nrg.2016.118. PMC 5206767. PMID 27795562.
- ^ Board on Life Sciences; Division on Earth and Life Studies; Committee on Science Technology; Policy and Global Affairs; Board on Health Sciences Policy; National Research Council; Institute of Medicine (13 April 2015). Gain-of-Function Research: Background and Alternatives. National Academies Press (US).
- ^ Eggertsson G, Adelberg EA (August 1965). «Map positions and specificities of suppressor mutations in Escherichia coli K-12». Genetics. 52 (2): 319–340. doi:10.1093/genetics/52.2.319. PMC 1210853. PMID 5324068.
- ^ Takiar V, Ip CK, Gao M, Mills GB, Cheung LW (March 2017). «Neomorphic mutations create therapeutic challenges in cancer». Oncogene. 36 (12): 1607–1618. doi:10.1038/onc.2016.312. PMC 6609160. PMID 27841866.
- ^ Ellis NA, Ciocci S, German J (February 2001). «Back mutation can produce phenotype reversion in Bloom syndrome somatic cells». Human Genetics. 108 (2): 167–73. doi:10.1007/s004390000447. PMID 11281456. S2CID 22290041.
- ^ Doolittle WF, Brunet TD (December 2017). «On causal roles and selected effects: our genome is mostly junk». BMC Biology. 15 (1): 116. doi:10.1186/s12915-017-0460-9. PMC 5718017. PMID 29207982.
- ^ Nichols RJ, Sen S, Choo YJ, Beltrao P, Zietek M, Chaba R, et al. (January 2011). «Phenotypic landscape of a bacterial cell». Cell. 144 (1): 143–56. doi:10.1016/j.cell.2010.11.052. PMC 3060659. PMID 21185072.
- ^ van Opijnen T, Bodi KL, Camilli A (October 2009). «Tn-seq: high-throughput parallel sequencing for fitness and genetic interaction studies in microorganisms». Nature Methods. 6 (10): 767–72. doi:10.1038/nmeth.1377. PMC 2957483. PMID 19767758.
- ^ Allen HL, Estrada K, Lettre G, Berndt SI, Weedon MN, Rivadeneira F, et al. (October 2010). «Hundreds of variants clustered in genomic loci and biological pathways affect human height». Nature. 467 (7317): 832–8. Bibcode:2010Natur.467..832L. doi:10.1038/nature09410. PMC 2955183. PMID 20881960.
- ^ Charlesworth D, Charlesworth B, Morgan MT (December 1995). «The pattern of neutral molecular variation under the background selection model». Genetics. 141 (4): 1619–32. doi:10.1093/genetics/141.4.1619. PMC 1206892. PMID 8601499.
- ^ Loewe L (April 2006). «Quantifying the genomic decay paradox due to Muller’s ratchet in human mitochondrial DNA». Genetical Research. 87 (2): 133–59. doi:10.1017/S0016672306008123. PMID 16709275.
- ^ Bernstein H, Hopf FA, Michod RE (1987). «The molecular basis of the evolution of sex». Molecular Genetics of Development. Advances in Genetics. Vol. 24. pp. 323–70. doi:10.1016/s0065-2660(08)60012-7. ISBN 9780120176243. PMID 3324702.
- ^ Peck JR, Barreau G, Heath SC (April 1997). «Imperfect genes, Fisherian mutation and the evolution of sex». Genetics. 145 (4): 1171–99. doi:10.1093/genetics/145.4.1171. PMC 1207886. PMID 9093868.
- ^ Simcikova D, Heneberg P (December 2019). «Refinement of evolutionary medicine predictions based on clinical evidence for the manifestations of Mendelian diseases». Scientific Reports. 9 (1): 18577. Bibcode:2019NatSR…918577S. doi:10.1038/s41598-019-54976-4. PMC 6901466. PMID 31819097.
- ^ Keightley PD, Lynch M (March 2003). «Toward a realistic model of mutations affecting fitness». Evolution; International Journal of Organic Evolution. 57 (3): 683–5, discussion 686–9. doi:10.1554/0014-3820(2003)057[0683:tarmom]2.0.co;2. JSTOR 3094781. PMID 12703958. S2CID 198157678.
- ^ Barton NH, Keightley PD (January 2002). «Understanding quantitative genetic variation». Nature Reviews Genetics. 3 (1): 11–21. doi:10.1038/nrg700. PMID 11823787. S2CID 8934412.
- ^ a b c Sanjuán R, Moya A, Elena SF (June 2004). «The distribution of fitness effects caused by single-nucleotide substitutions in an RNA virus». Proceedings of the National Academy of Sciences of the United States of America. 101 (22): 8396–401. Bibcode:2004PNAS..101.8396S. doi:10.1073/pnas.0400146101. PMC 420405. PMID 15159545.
- ^ Carrasco P, de la Iglesia F, Elena SF (December 2007). «Distribution of fitness and virulence effects caused by single-nucleotide substitutions in Tobacco Etch virus». Journal of Virology. 81 (23): 12979–84. doi:10.1128/JVI.00524-07. PMC 2169111. PMID 17898073.
- ^ Sanjuán R (June 2010). «Mutational fitness effects in RNA and single-stranded DNA viruses: common patterns revealed by site-directed mutagenesis studies». Philosophical Transactions of the Royal Society of London. Series B, Biological Sciences. 365 (1548): 1975–82. doi:10.1098/rstb.2010.0063. PMC 2880115. PMID 20478892.
- ^ Peris JB, Davis P, Cuevas JM, Nebot MR, Sanjuán R (June 2010). «Distribution of fitness effects caused by single-nucleotide substitutions in bacteriophage f1». Genetics. 185 (2): 603–9. doi:10.1534/genetics.110.115162. PMC 2881140. PMID 20382832.
- ^ Elena SF, Ekunwe L, Hajela N, Oden SA, Lenski RE (March 1998). «Distribution of fitness effects caused by random insertion mutations in Escherichia coli». Genetica. 102–103 (1–6): 349–58. doi:10.1023/A:1017031008316. PMID 9720287. S2CID 2267064.
- ^ a b Hietpas RT, Jensen JD, Bolon DN (May 2011). «Experimental illumination of a fitness landscape». Proceedings of the National Academy of Sciences of the United States of America. 108 (19): 7896–901. Bibcode:2011PNAS..108.7896H. doi:10.1073/pnas.1016024108. PMC 3093508. PMID 21464309.
- ^ Davies EK, Peters AD, Keightley PD (September 1999). «High frequency of cryptic deleterious mutations in Caenorhabditis elegans». Science. 285 (5434): 1748–51. doi:10.1126/science.285.5434.1748. PMID 10481013.
- ^ Loewe L, Charlesworth B (September 2006). «Inferring the distribution of mutational effects on fitness in Drosophila». Biology Letters. 2 (3): 426–30. doi:10.1098/rsbl.2006.0481. PMC 1686194. PMID 17148422.
- ^ Eyre-Walker A, Woolfit M, Phelps T (June 2006). «The distribution of fitness effects of new deleterious amino acid mutations in humans». Genetics. 173 (2): 891–900. doi:10.1534/genetics.106.057570. PMC 1526495. PMID 16547091.
- ^ Sawyer SA, Kulathinal RJ, Bustamante CD, Hartl DL (August 2003). «Bayesian analysis suggests that most amino acid replacements in Drosophila are driven by positive selection». Journal of Molecular Evolution. 57 (1): S154–64. Bibcode:2003JMolE..57S.154S. CiteSeerX 10.1.1.78.65. doi:10.1007/s00239-003-0022-3. PMID 15008412. S2CID 18051307.
- ^ Piganeau G, Eyre-Walker A (September 2003). «Estimating the distribution of fitness effects from DNA sequence data: implications for the molecular clock». Proceedings of the National Academy of Sciences of the United States of America. 100 (18): 10335–40. Bibcode:2003PNAS..10010335P. doi:10.1073/pnas.1833064100. PMC 193562. PMID 12925735.
- ^ Kimura M (February 1968). «Evolutionary rate at the molecular level». Nature. 217 (5129): 624–6. Bibcode:1968Natur.217..624K. doi:10.1038/217624a0. PMID 5637732. S2CID 4161261.
- ^ Akashi H (September 1999). «Within- and between-species DNA sequence variation and the ‘footprint’ of natural selection». Gene. 238 (1): 39–51. doi:10.1016/S0378-1119(99)00294-2. PMID 10570982.
- ^ Eyre-Walker A (October 2006). «The genomic rate of adaptive evolution». Trends in Ecology & Evolution. 21 (10): 569–75. doi:10.1016/j.tree.2006.06.015. PMID 16820244.
- ^ Gillespie JH (September 1984). «Molecular Evolution Over the Mutational Landscape». Evolution. 38 (5): 1116–1129. doi:10.2307/2408444. JSTOR 2408444. PMID 28555784.
- ^ Orr HA (April 2003). «The distribution of fitness effects among beneficial mutations». Genetics. 163 (4): 1519–26. doi:10.1093/genetics/163.4.1519. PMC 1462510. PMID 12702694.
- ^ Kassen R, Bataillon T (April 2006). «Distribution of fitness effects among beneficial mutations before selection in experimental populations of bacteria». Nature Genetics. 38 (4): 484–8. doi:10.1038/ng1751. PMID 16550173. S2CID 6954765.
- ^ Rokyta DR, Joyce P, Caudle SB, Wichman HA (April 2005). «An empirical test of the mutational landscape model of adaptation using a single-stranded DNA virus». Nature Genetics. 37 (4): 441–4. doi:10.1038/ng1535. PMID 15778707. S2CID 20296781.
- ^ Imhof M, Schlotterer C (January 2001). «Fitness effects of advantageous mutations in evolving Escherichia coli populations». Proceedings of the National Academy of Sciences of the United States of America. 98 (3): 1113–7. Bibcode:2001PNAS…98.1113I. doi:10.1073/pnas.98.3.1113. PMC 14717. PMID 11158603.
- ^ a b «Somatic cell genetic mutation». Genome Dictionary. Athens, Greece: Information Technology Associates. 30 June 2007. Archived from the original on 24 February 2010. Retrieved 6 June 2010.
- ^ «Compound heterozygote». MedTerms. New York: WebMD. 14 June 2012. Archived from the original on 4 March 2016. Retrieved 9 October 2015.
- ^ «RB1 Genetics». Daisy’s Eye Cancer Fund. Oxford, UK. Archived from the original on 26 November 2011. Retrieved 9 October 2015.
- ^ Cotton, RG (1993). «Current methods of mutation detection». Mutat Res. 285 (1): 125–144. doi:10.1016/0027-5107(93)90060-S. PMID 7678126.
- ^ a b Lerman, LS; Silverstein, K (1987). «Computational simulation of DNA melting and its application to denaturing gradient gel electrophoresis». Methods Enzymol. 155: 482–501. doi:10.1016/0076-6879(87)55032-7. PMID 2828875.
- ^ «somatic mutation | genetics». Encyclopædia Britannica. Archived from the original on 31 March 2017. Retrieved 31 March 2017.
- ^ Hartl L, Jones EW (1998). Genetics Principles and Analysis. Sudbury, Massachusetts: Jones and Bartlett Publishers. pp. 556. ISBN 978-0-7637-0489-6.
- ^ Milholland B, Dong X, Zhang L, Hao X, Suh Y, Vijg J (May 2017). «Differences between germline and somatic mutation rates in humans and mice». Nature Communications. 8: 15183. Bibcode:2017NatCo…815183M. doi:10.1038/ncomms15183. PMC 5436103. PMID 28485371.
- ^ Alberts B (2014). Molecular Biology of the Cell (6 ed.). Garland Science. p. 487. ISBN 9780815344322.
- ^ a b Chadov BF, Fedorova NB, Chadova EV (1 July 2015). «Conditional mutations in Drosophila melanogaster: On the occasion of the 150th anniversary of G. Mendel’s report in Brünn». Mutation Research/Reviews in Mutation Research. 765: 40–55. doi:10.1016/j.mrrev.2015.06.001. PMID 26281767.
- ^ a b Landis G, Bhole D, Lu L, Tower J (July 2001). «High-frequency generation of conditional mutations affecting Drosophila melanogaster development and life span». Genetics. 158 (3): 1167–76. doi:10.1093/genetics/158.3.1167. PMC 1461716. PMID 11454765. Archived from the original on 22 March 2017. Retrieved 21 March 2017.
- ^ a b c d Gierut JJ, Jacks TE, Haigis KM (April 2014). «Strategies to achieve conditional gene mutation in mice». Cold Spring Harbor Protocols. 2014 (4): 339–49. doi:10.1101/pdb.top069807. PMC 4142476. PMID 24692485.
- ^ Spencer DM (May 1996). «Creating conditional mutations in mammals». Trends in Genetics. 12 (5): 181–7. doi:10.1016/0168-9525(96)10013-5. PMID 8984733.
- ^ Tan G, Chen M, Foote C, Tan C (September 2009). «Temperature-sensitive mutations made easy: generating conditional mutations by using temperature-sensitive inteins that function within different temperature ranges». Genetics. 183 (1): 13–22. doi:10.1534/genetics.109.104794. PMC 2746138. PMID 19596904.
- ^ den Dunnen JT, Antonarakis SE (January 2000). «Mutation nomenclature extensions and suggestions to describe complex mutations: a discussion». Human Mutation. 15 (1): 7–12. doi:10.1002/(SICI)1098-1004(200001)15:1<7::AID-HUMU4>3.0.CO;2-N. PMID 10612815. S2CID 84706224.
- ^ Jónsson H, Sulem P, Kehr B, Kristmundsdottir S, Zink F, Hjartarson E, et al. (September 2017). «Parental influence on human germline de novo mutations in 1,548 trios from Iceland». Nature. 549 (7673): 519–522. Bibcode:2017Natur.549..519J. doi:10.1038/nature24018. PMID 28959963. S2CID 205260431.
- ^ «Study challenges evolutionary theory that DNA mutations are random». U.C. Davis. Retrieved 12 February 2022.
- ^ Monroe JG, Srikant T, Carbonell-Bejerano P, Becker C, Lensink M, Exposito-Alonso M, et al. (February 2022). «Mutation bias reflects natural selection in Arabidopsis thaliana». Nature. 602 (7895): 101–105. Bibcode:2022Natur.602..101M. doi:10.1038/s41586-021-04269-6. PMC 8810380. PMID 35022609.
- ^ Doniger SW, Kim HS, Swain D, Corcuera D, Williams M, Yang SP, Fay JC (August 2008). Pritchard JK (ed.). «A catalog of neutral and deleterious polymorphism in yeast». PLOS Genetics. 4 (8): e1000183. doi:10.1371/journal.pgen.1000183. PMC 2515631. PMID 18769710.
- ^ Ionov Y, Peinado MA, Malkhosyan S, Shibata D, Perucho M (June 1993). «Ubiquitous somatic mutations in simple repeated sequences reveal a new mechanism for colonic carcinogenesis». Nature. 363 (6429): 558–61. Bibcode:1993Natur.363..558I. doi:10.1038/363558a0. PMID 8505985. S2CID 4254940.
- ^ Araten DJ, Golde DW, Zhang RH, Thaler HT, Gargiulo L, Notaro R, Luzzatto L (September 2005). «A quantitative measurement of the human somatic mutation rate». Cancer Research. 65 (18): 8111–7. doi:10.1158/0008-5472.CAN-04-1198. PMID 16166284.
- ^ «‘Lifeless’ prion proteins are ‘capable of evolution’«. Health. BBC News. London. 1 January 2010. Archived from the original on 25 September 2015. Retrieved 10 October 2015.
- ^ Sullivan AD, Wigginton J, Kirschner D (August 2001). «The coreceptor mutation CCR5Delta32 influences the dynamics of HIV epidemics and is selected for by HIV». Proceedings of the National Academy of Sciences of the United States of America. 98 (18): 10214–9. Bibcode:2001PNAS…9810214S. doi:10.1073/pnas.181325198. PMC 56941. PMID 11517319.
- ^ «Mystery of the Black Death». Secrets of the Dead. Season 3. Episode 2. 30 October 2002. PBS. Archived from the original on 12 October 2015. Retrieved 10 October 2015. Episode background.
- ^ Galvani AP, Slatkin M (December 2003). «Evaluating plague and smallpox as historical selective pressures for the CCR5-Delta 32 HIV-resistance allele». Proceedings of the National Academy of Sciences of the United States of America. 100 (25): 15276–9. Bibcode:2003PNAS..10015276G. doi:10.1073/pnas.2435085100. PMC 299980. PMID 14645720.
- ^ Konotey-Ahulu F. «Frequently Asked Questions [FAQ’s]». sicklecell.md. Archived from the original on 30 April 2011. Retrieved 16 April 2010.
- ^ Hughes D, Andersson DI (September 2017). «Evolutionary Trajectories to Antibiotic Resistance». Annual Review of Microbiology. 71: 579–596. doi:10.1146/annurev-micro-090816-093813. PMID 28697667.
- ^ Ségurel L, Bon C (August 2017). «On the Evolution of Lactase Persistence in Humans». Annual Review of Genomics and Human Genetics. 18: 297–319. doi:10.1146/annurev-genom-091416-035340. PMID 28426286.
- ^ a b c Barešić, Anja; Martin, Andrew C.R. (1 August 2011). «Compensated pathogenic deviations». BioMolecular Concepts. 2 (4): 281–292. doi:10.1515/bmc.2011.025. ISSN 1868-503X. PMID 25962036. S2CID 6540447.
- ^ a b c Whitlock, Michael C.; Griswold, Cortland K.; Peters, Andrew D. (2003). «Compensating for the meltdown: The critical effective size of a population with deleterious and compensatory mutations». Annales Zoologici Fennici. 40 (2): 169–183. ISSN 0003-455X. JSTOR 23736523.
- ^ a b Lanfear, Robert; Kokko, Hanna; Eyre-Walker, Adam (1 January 2014). «Population size and the rate of evolution». Trends in Ecology & Evolution. 29 (1): 33–41. doi:10.1016/j.tree.2013.09.009. ISSN 0169-5347. PMID 24148292.
- ^ Doudna, Jennifer A. (1 November 2000). «Structural genomics of RNA». Nature Structural Biology. 7: 954–956. doi:10.1038/80729. PMID 11103998. S2CID 998448.
- ^ Cowperthwaite, Matthew C.; Bull, J. J.; Meyers, Lauren Ancel (20 October 2006). «From Bad to Good: Fitness Reversals and the Ascent of Deleterious Mutations». PLOS Computational Biology. 2 (10): e141. Bibcode:2006PLSCB…2..141C. doi:10.1371/journal.pcbi.0020141. ISSN 1553-7358. PMC 1617134. PMID 17054393.
- ^ Cowperthwaite, Matthew C.; Meyers, Lauren Ancel (1 December 2007). «How Mutational Networks Shape Evolution: Lessons from RNA Models». Annual Review of Ecology, Evolution, and Systematics. 38 (1): 203–230. doi:10.1146/annurev.ecolsys.38.091206.095507. ISSN 1543-592X.
- ^ a b Azbukina, Nadezhda; Zharikova, Anastasia; Ramensky, Vasily (1 October 2022). «Intragenic compensation through the lens of deep mutational scanning». Biophysical Reviews. 14 (5): 1161–1182. doi:10.1007/s12551-022-01005-w. ISSN 1867-2469. PMC 9636336. PMID 36345285.
- ^ a b c d DePristo, Mark A.; Weinreich, Daniel M.; Hartl, Daniel L. (September 2005). «Missense meanderings in sequence space: a biophysical view of protein evolution». Nature Reviews. Genetics. 6 (9): 678–687. doi:10.1038/nrg1672. ISSN 1471-0056. PMID 16074985. S2CID 13236893.
- ^ a b Ferrer-Costa, Carles; Orozco, Modesto; Cruz, Xavier de la (5 January 2007). «Characterization of Compensated Mutations in Terms of Structural and Physico-Chemical Properties». Journal of Molecular Biology. 365 (1): 249–256. doi:10.1016/j.jmb.2006.09.053. ISSN 0022-2836. PMID 17059831.
- ^ Lunzer, Mark; Golding, G. Brian; Dean, Antony M. (21 October 2010). «Pervasive Cryptic Epistasis in Molecular Evolution». PLOS Genetics. 6 (10): e1001162. doi:10.1371/journal.pgen.1001162. ISSN 1553-7404. PMC 2958800. PMID 20975933.
- ^ a b Corrigan, Rebecca M.; Abbott, James C.; Burhenne, Heike; Kaever, Volkhard; Gründling, Angelika (1 September 2011). «c-di-AMP Is a New Second Messenger in Staphylococcus aureus with a Role in Controlling Cell Size and Envelope Stress». PLOS Pathogens. 7 (9): e1002217. doi:10.1371/journal.ppat.1002217. ISSN 1553-7366. PMC 3164647. PMID 21909268.
- ^ a b c Comas, Iñaki; Borrell, Sonia; Roetzer, Andreas; Rose, Graham; Malla, Bijaya; Kato-Maeda, Midori; Galagan, James; Niemann, Stefan; Gagneux, Sebastien (January 2012). «Whole-genome sequencing of rifampicin-resistant Mycobacterium tuberculosis strains identifies compensatory mutations in RNA polymerase genes». Nature Genetics. 44 (1): 106–110. doi:10.1038/ng.1038. ISSN 1546-1718. PMC 3246538. PMID 22179134.
- ^ a b Gagneux, Sebastien; Long, Clara Davis; Small, Peter M.; Van, Tran; Schoolnik, Gary K.; Bohannan, Brendan J. M. (30 June 2006). «The competitive cost of antibiotic resistance in Mycobacterium tuberculosis». Science. 312 (5782): 1944–1946. doi:10.1126/science.1124410. ISSN 1095-9203. PMID 16809538. S2CID 7454895.
- ^ a b c Reynolds, M. G. (December 2000). «Compensatory evolution in rifampin-resistant Escherichia coli». Genetics. 156 (4): 1471–1481. doi:10.1093/genetics/156.4.1471. ISSN 0016-6731. PMC 1461348. PMID 11102350.
- ^ Gong, Lizhi Ian; Suchard, Marc A; Bloom, Jesse D (14 May 2013). Pascual, Mercedes (ed.). «Stability-mediated epistasis constrains the evolution of an influenza protein». eLife. 2: e00631. doi:10.7554/eLife.00631. ISSN 2050-084X. PMC 3654441. PMID 23682315.
- ^ a b Davis, Brad H.; Poon, Art F.Y.; Whitlock, Michael C. (22 May 2009). «Compensatory mutations are repeatable and clustered within proteins». Proceedings of the Royal Society B: Biological Sciences. 276 (1663): 1823–1827. doi:10.1098/rspb.2008.1846. ISSN 0962-8452. PMC 2674493. PMID 19324785.
- ^ Azbukina, Nadezhda; Zharikova, Anastasia; Ramensky, Vasily (1 October 2022). «Intragenic compensation through the lens of deep mutational scanning». Biophysical Reviews. 14 (5): 1161–1182. doi:10.1007/s12551-022-01005-w. ISSN 1867-2469. PMC 9636336. PMID 36345285.
- ^ a b Burch, Christina L; Chao, Lin (1 March 1999). «Evolution by Small Steps and Rugged Landscapes in the RNA Virus ϕ6». Genetics. 151 (3): 921–927. doi:10.1093/genetics/151.3.921. ISSN 1943-2631. PMC 1460516. PMID 10049911.
- ^ a b Rimmelzwaan, G. F.; Berkhoff, E. G. M.; Nieuwkoop, N. J.; Smith, D. J.; Fouchier, R. A. M.; Osterhaus, A. D. M. E.YR 2005 (2005). «Full restoration of viral fitness by multiple compensatory co-mutations in the nucleoprotein of influenza A virus cytotoxic T-lymphocyte escape mutants». Journal of General Virology. 86 (6): 1801–1805. doi:10.1099/vir.0.80867-0. ISSN 1465-2099. PMID 15914859.
- ^ Kimura, Motoo (1 July 1985). «The role of compensatory neutral mutations in molecular evolution». Journal of Genetics. 64 (1): 7–19. doi:10.1007/BF02923549. ISSN 0973-7731. S2CID 129866.
- ^ a b c Bowler PJ (1992) [1983]. The Eclipse of Darwinism. p. 198.
- ^ Smocovitis VB (1996). «Unifying biology: the evolutionary synthesis and evolutionary biology». Journal of the History of Biology. Princeton University Press. 25 (1): 1–65. doi:10.1007/bf01947504. ISBN 978-0-691-03343-3. LCCN 96005605. OCLC 34411399. PMID 11623198. S2CID 189833728.
- ^ Hallgrímsson B, Hall BK (2011). Variation: A Central Concept in Biology. Academic Press. p. 18.
- ^ Sewall Wright. (1984). Evolution and the Genetics of Populations: Genetics and Biometric Foundations Volume 1. University of Chicago Press. p. 10
- ^ De Vries H (1905). Species and Varieties: Their Origin by Mutation.
- ^ Bateson W (1894). Materials for the Study of Variation, Treated with Especial Regard to Discontinuity in the Origin of Species.
- ^ Punnett RC (1915). Mimicry in Butterflies. Cambridge University Press.
- ^ Mayr E (2007). What Makes Biology Unique?: Considerations on the Autonomy of a Scientific Discipline. Cambridge University Press.
- ^ a b Provine WB (2001). The Origins of Theoretical Population Genetics, with a new afterword. University of Chicago Press, Chicago. pp. 56–107.
- ^ a b Stoltzfus A, Cable K (2014). «Mendelian-mutationism: the forgotten evolutionary synthesis». Journal of the History of Biology. 47 (4): 501–46. doi:10.1007/s10739-014-9383-2. PMID 24811736. S2CID 23263558.
- ^ Hull DL (1985). «Darwinism as an historical entity: A historiographic proposal». In Kohn D (ed.). The Darwinian Heritage. Princeton University Press. pp. 773–812. ISBN 9780691024141.
- ^ Yule GU (1902). «Mendel’s Laws and their probable relations to inter-racial heredity». New Phytologist. 1 (10): 226–227. doi:10.1111/j.1469-8137.1902.tb07336.x.
- ^ Gould SJ (1982). The uses of heresey; an introduction to Richard Goldschmidt’s The Material Basis of Evolution. Yale University Press. pp. xiii–xlii. ISBN 0300028237.
- ^ Ruse M (1996). Monad to man: the Concept of Progress in Evolutionary Biology. Harvard University Press. pp. 412–413. ISBN 978-0-674-03248-4.
- ^ Stoltzfus A (2014). «In search of mutation-driven evolution». Evolution & Development. 16: 57–59. doi:10.1111/ede.12062.
- ^ Futuyma DJ (2015). Serrelli E, Gontier N (eds.). Can Modern Evolutionary Theory Explain Macroevolution? (PDF). Macroevolution. Springer. pp. 29–85. Archived from the original (PDF) on 23 February 2020. Retrieved 31 October 2017.
External links[edit]
![]()
Wikimedia Commons has media related to Mutations.
- Jones S, Woolfson A, Partridge L (6 December 2007). «Genetic Mutation». In Our Time. BBC Radio 4. Retrieved 18 October 2015.
- Liou S (5 February 2011). «All About Mutations». HOPES. Huntington’s Disease Outreach Project for Education at Stanford. Retrieved 18 October 2015.
- «Locus Specific Mutation Databases». Leiden, the Netherlands: Leiden University Medical Center. Retrieved 18 October 2015.
- «Welcome to the Mutalyzer website». Leiden, the Netherlands: Leiden University Medical Center. Retrieved 18 October 2015. – The Mutalyzer website.
Генетические мутации — изменения в генах, которые вызывают неизлечимые заболевания. Но все ли мутации опасны? И почему такое вообще случается? Если у человека есть генетическая мутация, значит ли это, что у него проявится какая-то болезнь? Отвечаем на эти и другие вопросы в статье.
Содержание
- Что такое ДНК?
- Какова функция генов?
- Что такое мутация?
- Мутации: хорошие, плохие, нейтральные
- Что делать, если генетический тест показал мутацию?
Что такое ДНК?
Чтобы разобраться, что такое генетическая мутация, вспомним, как устроены ДНК и гены.
ДНК (дезоксирибонуклеиновая кислота) — это длинная молекула, которую принято называть «двойной спиралью». Она хранит биологическую информацию, которая «записана» в виде генетического кода.
Ген — это основная «единица» наследственной информации. Он представляет собой кусочек ДНК.
Какова функция генов?
В части генов в виде кода записаны «рецепты» изготовления белков. Именно белки выполняют основные функции для поддержания жизнедеятельности организма: они отвечают за пищеварение, кровообращение, иммунитет, передачу информации между клетками.
Код представляет собой последовательность нуклеотидов.
Нуклеотид — это конструкция, которая состоит из молекулы сахара, молекулы фосфата и основания.
В нашей ДНК есть четыре азотистых основания:
- аденин,
- гуанин,
- цитозин,
- тимин.
Основания одной цепи соединяются с основаниями другой цепи парами (аденин с тимином, цитозин с гуанином).
Если посмотреть на двойную спираль ДНК, то ее горизонтальные «ступени» будут парами оснований, а вертикальные боковые части — сахарами и фосфатами.
Чтобы изготовить белки по записанному в генах коду, специальные соединения — ферменты — «читают» и копируют код. В результате получаются длинные одноцепочечные молекулы — РНК (рибонуклеиновые кислоты), но это еще не белок. РНК лишь несут в себе информацию о первичной структуре белка, поэтому их называют матричными (сокращенно — мРНК). Эти молекулы покидают ядро клетки и перемещаются в ее цитоплазму. Там специальные органы — рибосомы — считывают код мРНК и изготавливают по этому «рецепту» белок.
Что такое мутация?
Генетическая мутация — это любое изменение в нуклеотидной последовательности ДНК.
К основным типам мутаций относятся:
- транзиция — замена аденина на гуанин или замена тимина на цитозин;
- трансверсия — аденин или гуанин меняются местами с тимином или цитозином;
- делеция — потеря участка ДНК;
- инсерция — добавление участка ДНК;
- дупликация — удвоение участка ДНК;
- инверсия — изменение, при котором участок хромосомы поворачивается на 180°;
- транслокация — мутация, при которой хромосомы обмениваются фрагментами.
Мутации могут происходить по разным причинам.
Спонтанные генетические мутации
Они происходят на протяжении всей нашей жизни. Можно сказать, что это нормальное явление, которое случается в ходе разных процессов, например, при копировании ДНК.
Как правило, такие ошибки не грозят серьезными последствиями, потому что у нашего организма есть механизмы защиты.
К ним относится, например, апоптоз — процесс программируемой гибели «испорченной» клетки, или репарация — починка нити ДНК. В этом случае ошибочный участок ДНК вырезается, а на его месте формируется новый.
Мутации, вызванные внешним влиянием
Мутации могут возникнуть под воздействием внешних неблагоприятных факторов, например, химических веществ, ионизирующего излучения или заражения вирусами.
Белки, которые отвечают за исправление ошибок, как правило, могут исправить испорченные цепи ДНК или привести одну хромосому в соответствие с другой. Но, если ошибки произошли на уровне генома или количества хромосом, защитные механизмы будут бессильны.
Наследственные генетические мутации
Такие мутации достаются человеку от родителей. Бывают случаи, когда генетическое нарушение передается из поколения в поколение (как, например, болезнь гемофилия), иногда мутации происходят в яйцеклетках и сперматозоидах и таким образом передаются ребенку.
Бывают случаи, когда мутации возникают на этапе формирования зиготы — клетки, которая образуется в результате оплодотворения. Как и в предыдущем случае, механизмы репарации с такими мутациями работают далеко не всегда, а ряд заболеваний и вовсе связан с нарушениями в процессе починки (например, пигментная ксеродерма — заболевание кожи, представляющее собой повышенную чувствительность к ультрафиолету).
Мутации: хорошие, плохие, нейтральные
Не все генетические мутации опасны. Важно понимать, что именно мутации объясняют генетические различия между видами. Изменения генов влекут за собой изменение характеристик организма, и в результате этого он может стать либо более, либо менее приспособленным к выживанию.
В ходе естественного отбора преимущество получают те живые существа, которые обладают более «полезным» набором характеристик, и тогда мутация закрепляется в популяции, становясь нормой.
«Хорошие» мутации
Ученым известно, что, например, у людей с определенным вариантом гена GPR75 риск ожирения снижен на 54%. А те, у кого есть хотя бы одна копия такого варианта гена, имеют более низкий индекс массы тела.
Мутации генов могут давать человеку и другие преимущества: так, мутировавший ген EPOR дал финскому лыжнику, трехкратному олимпийскому чемпиону Ээро Мянтюранта высокую чувствительность к эритропоэтину — гормону, который помогает нашим клеткам поддерживать оптимальный уровень кислорода и выводить углекислый газ. Это изменило и объем красных кровяных клеток в крови спортсмена, и объем кислорода, который эти клетки способны переносить. В результате Мянтюранта получил супервыносливость — его организм легко справлялся с повышенной потребностью в кислороде во время физических нагрузок.
«Плохие» мутации
Генетические мутации могут вызывать различные заболевания. Например, изменения гена DMD вызывают дистрофию Дюшенна — нервно-мышечное заболевание, которое проявляется у мужчин намного чаще, чем у женщин. А к серповидноклеточной анемии — нарушению в строении белка гемоглобина, который переносит кислород от легких к органам, — приводят мутации гена HBB. Хорея Гентингтона — тяжелое заболевание нервной системы — развивается из-за мутации в гене HTT.
Однако далеко не всегда генетическое заболевание связано с мутацией одного гена. Так, синдром Дауна возникает из-за изменения количества хромосом — в клетках пациентов с этой болезнью 47 хромосом вместо обычных 46.
Ряд заболеваний, таких как рак, диабет, расстройства аутического спектра, появляются из-за комбинации факторов. Пациенты могут иметь генетическую предрасположенность, но значительную роль играют и внешние факторы — неправильный образ жизни, неблагоприятная окружающая среда.
«Нейтральные» мутации
Нейтральные мутации, как следует из названия, не оказывают на здоровье человека ни положительного, ни отрицательного эффекта. Как правило, эти мутации затрагивают нуклеотидную последовательность ДНК, но не сказываются на строении белков.
Так происходит, потому что наш генетический код обладает так называемой избыточностью — это значит, что ряд аминокислот кодируется несколькими способами, чтобы случайные ошибки при копировании с меньшей вероятностью привели к нарушению функции или отсутствию кодируемого белка.
Бывает и так, что мутация гена все-таки меняет аминокислоту. Тем не менее, это не всегда приводит к нарушению функции белка.
Что делать, если генетический тест показал мутацию?
Во многих случаях наличие генетических нарушений не означает, что у человека непременно разовьется какое-то заболевание. Это значит лишь то, что пациент обладает генетическим вариантом, который чаще встречается у людей с этим заболеванием, и, вероятно, предрасположен к возникновению проблемы больше остальных.
Важнейшую роль в таких случаях играют внешние факторы: образ жизни, привычки, окружающая среда.
Ученые сходятся на том, что важными условиями сохранения здоровья являются:
● сбалансированное питание, богатое овощами и фруктами;
● регулярные занятия спортом;
● отказ от курения и алкоголя;
● достаточное количество сна.
Эти правила помогут значительно снизить риск развития таких распространенных заболеваний, как рак, проблемы с сердечно-сосудистой системой, диабет второго типа.
В том случае, если у человека есть мутация, связанная с моногенным заболеванием (то есть таким, которое возникает из-за «поломки» всего лишь одного гена), то существует риск, что он передаст этот вариант гена своему ребенку. Кроме того, болезнь может проявиться и у самого обладателя «плохого» гена — в этом случае ему следует обратиться к специалистам. Как правило, генетические заболевания не лечатся, но врач сможет порекомендовать препараты или изменения образа жизни (например, диета), чтобы уменьшить проявления болезни.
Результаты Генетического теста Атлас подскажут персональные рекомендации по улучшению образа жизни, которые помогут минимизировать риск появления заболеваний. Используя эти знания, будет проще спланировать подходящий рацион, спортивные нагрузки и тренировки, профилактические обследования.
Больше статей о генетике в блоге Атласа:
- Из чего состоит геном человека
- Кому и зачем нужны ДНК-банки
- Генная терапия: шанс или фантастика
- Very Well Health, What Are Genes, DNA, and Chromosomes? 2022
- Your Genome, What is a genome? 2017
- Medline Plus, What is DNA? 2021
- National Human Genome Research Institute, Mutation