Причины мутаций
Точечные
мутации, суть которых связана с заменами
одной пары оснований на другую пару,
могут быть вызваны самыми различными
причинами. К числу наиболее известных
и хорошо изученных факторов, вызывающих
появление точечных мутаций относятся
нижеследующие.
Ошибки
репликации, не исправленные ДНК-полимеразой.
Как известно, точность копирования в
процессе репликации ДНК настолько
велика, что в среднем на каждые 1 10 9
пар нуклеотидов приходится одна
ошибка. Такую высокую точность репликации
обеспечивает корректирующая (3′
5′)-экзонуклеазная активность
ДНК-полимеразы. Тем не менее, в ряде
случаев ДНК-полимераза может ошибаться.
Одной из причин таких ошибок является
способность всех азотистых оснований
образовывать термодинамически невыгодные
таутомерные формы за счет миграции
атома водорода. При этом амино- и
оксогруппы превращаются в иминогруппы
(=NH) и енольные группы
(-ОН), соответственно. Такие редкие
таутомерные формы, как правило, образуют
неправильные, неканонические пары с
другими основаниями. Примером может
служить способность Cyt образовывать
редкую таутомерную имино-форму,
приведенную на рис 5.4.

|
Рис. 5.4 |
Таутомерная |
Эта
имино-форма
Cyt
образует пару не с Gua, а с Ade. В результате,
в процессе последующей репликации,
может произойти замена пары A-T на G-C.
Точно также аденин способен образовывать
редкую таутомерную имино-форму, которая
приобретает способность комплементарно
спариваться с неканоническим для него
цитозином (рис. 5.5).
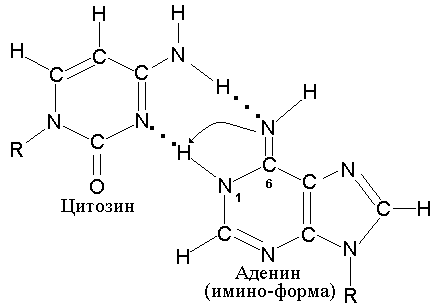
|
Рис. 5.5 |
Таутомерная |
Процессы
спонтанного дезаминирования обычных
и модифицированных оснований. В
настоящее время хорошо известно, что
некоторая часть оснований, входящих в
состав ДНК как про-, так и эукариот
метилируется пострепликативно особыми
ферментами — метилазами.
Цели метилирования у про- и эукариот
различны, но эта модификация ДНК протекает
достаточно интенсивно. Чаще всего
метилированию подвергаются остатки
Ade и Cyt. Ранее нами была рассмотрена
схема, показывающая последовательность
событий приводящих к возникновению
транзиции G-C
A-T
вследствие спонтанного или индуцированного
дезаминирования цитозина (рис. 5.1), а
также причины существования горячих
точек.
Например,
если дезаминированию подвергается не
цитозин, а 5-метилцитозин,
то этот процесс приводит к образованию
нормального для днк
основания – тимина (Thy). Естественно,
что в этом случае при повторной репликации
возможна замена пары G-C на пару A-T. Было
обнаружено, что процессы спонтанного
дезаминирования происходят с определенной
достаточно высокой скоростью, которая
составляет 100 актов дезаминирования на
один геном в сутки.
Процессы
апуринизации ДНК.
ДНК каждой клетки человеческого организма
в результате апуринизации
теряет в сутки около 5.000 пуриновых
оснований (Ade и Gua) вследствие термального
разрыва N-гликозидных связей между
пуриновым основанием и дезоксирибозой.
Удаление пиримидиновых оснований из
ДНК в какой-либо ощутимой степени не
происходит из-за того, что N-гликозидные
связи пиримидинов с углеводной частью
намного более стабильны, чем связь
пурина с углеводом. На интенсивность
процесса апуринизации ДНК могут влиять
также различные химические факторы.
Например, в кислой среде эффективность
апуринизации ДНК существенно возрастает.
Алкилирование гуанина под действием
диметилсульфата приводит к образованию
четвертичного азота в 7-ом положении
этого азотистого основания, что ослабляет
N-гликозидную связь с дезоксирибозой и
высвобождает метильное производное
гуанина, обеспечивая дополнительную
потерю пуринов молекулами ДНК (рис.
5.6).


.
|
Рис. 5.6 |
Алкилирующий |
Повреждения
ДНК, вызываемые действием химических
факторов окружающей среды.
Основания в составе ДНК весьма
чувствительны к действию многочисленных
химических соединений, распространенных
в окружающей среде. Многие из них получили
название ксенобиотиков, большинство
которых имеет антропогенное техногенное
происхождение. К разряду ксенобиотиков
относятся многочисленные яды, лекарства,
канцерогены, пестициды, инсектициды,
гербициды и многие другие соединения.
Среди эффектов различных соединений
наиболее полно изучено воздействие
азотистой кислоты (HNO2)
(рис. 5.3), гидроксиламина (NH2OH)
(рис.5.7), алкилирующих агентов, таких
как диметилсульфат (рис. 5.6),
N-метил-N/-нитро-N-нитрозогуанидин:
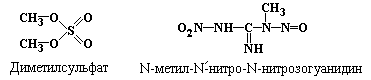

.
|
Рис. 5.7 |
Реакция цитозина |
Под
действием азотистой
кислоты,
которая может образовываться из таких
предшественников как NaNO2
— нитрит натрия, NaNO3
— нитрат натрия, а также органических
соединений типа нитрозаминов

происходит
активное дезаминирование Cyt с образованием
Ura, Ade с образованием гипоксантина (Hyp) и
Gua с образованием ксантина (Xan). В
результате дезаминирования Cyt образуется,
как уже известно, Ura, который комплементарно
спаривается с Ade. В результате происходит
транзиция G-C
A-T.
С
другой стороны, в результате дезаминирования
Ade образуется Hyp, который приобретает
способность спариваться с Cyt, вызывая
транзицию A-T
G-C. если
события, касающиеся дезаминирования,
произойдут в одном сайте, т.е. сначала
дезаминированию подвергнется Cyt и
произойдет образование A-T пары вместо
G-C, а затем дезаминированию подвергнется
появившийся Ade и произойдет обратная
транзиция A-T
G-C, то при этом последовательность ДНК
восстановится.
Дезаминирование
Gua в Xan не влияет на способность измененного
основания образовывать пару с цитозином
(Xan – Cyt).
Алкилирующие
агенты могут
воздействовать как на структуру
оснований, так могут разрушать и
фосфодиэфирные связи, приводя к
фрагментированию цепей ДНК. Кроме того,
некоторые алкилирующие агенты способны
ковалентно взаимодействовать с обеими
цепями ДНК, вызывая образование поперечных
сшивок.
Кроме
упомянутого выше диметилсульфата, к
числу наиболее активных алкилирующих
агентов относятся
диметилнитрозамин и
азотистый
иприт:
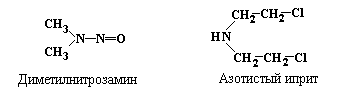
В результате
воздействия алкилирующих (как метилирующих,
так и этилирующих) агентов может
происходить метилирование Gua по
7-положению, что приводит к образованию
7-метилгуанина (рис. 5.8), который впоследствие
образует неканоническую пару с тимином.
С тимином также может спариваться
7-этилгуанин.

|
Рис. 5.8 |
Взаимодействие |
Образование
неканонической пары между тимином и
7-этилгуанином показано на рис. 5.9.

|
Рис. 5.9 |
Характер |
Кроме
приведенных выше способов модификации
гуанина, это азотистое основание может
также метилироваться по гидроксильной
группе енольной формы с образованием
O6-метил-Gua,
не способного образовывать нормальную
комплементарную пару с Cyt. В свою очередь,
метилирование аденина по аминогруппе
приводит к образованию N6-метиладенина.
Ошибки
ДНК-полимеразы,
связанные
с
включением
аналогов природных нуклеотидов.
Одним из аналогов природных нуклеотидов,
не выщепляемых ДНК-полимеразой является
нуклеотид с азотистым основанием —
2-аминопурином (рис. 5.10), который
встраивается в ДНК вместо Ade, но
впоследствии спаривается с цитозином
и тем самым способствует транзиции A-T
G-C.

|
Рис. 5.10 |
Аналог аденина |
Несмотря
на высокую точность функционирования
ДНК-полимераз при катализе репликации
эти ферменты не всегда способны отличать
нормальные дезоксирибонуклеозид
трифосфатные субстраты от некоторых
других нуклеотидов с очень похожей
структурой. Следует отметить, что в
случае 2-аминопурина ДНК-полимераза все
же делает существенную ошибку, поскольку
наличие NH2-группы
во 2-ом или в 6-ом положениях пурина
структурно является очень заметным. На
рис. 5.11, приведенном ниже, представлена
схема возникновения транзиции A-T
G-C, инициируемая встраиванием 2-аминопурина.

|
Рис. 5.11 |
Схема, отражающая |
Другой
пример таких структурных аналогов
природных нуклеозидтрифосфатов –
5-Br-dUTP
(5-Br-дезоксиуридинтрифосфат),
который является аналогом тимидинтрифосфата
из-за присутствия атома Br в 5-положении
урацила, где у тимина находится CH3-группа.
Первоначально 5-галоидпроизводные
урацила были синтезированы как аналоги
тимина с целью их возможного применения
в качестве цитостатиков или противовирусных
средств. В частности цитостатические
эффекты 5-Br-dUrd сводятся к эффективному
фосфорилированию данного аналога
тимидина под действием тимидинкиназы
поврежденной клетки и встраиванию этого
модифицированного нуклеотида в ДНК. В
результате последующего облучения
клеток ультрафиолетовым светом встроенный
5-Br-Ura принимает участие в образовании
большого числа пиримидиновых димеров
и поперечных сшивок в ДНК, что практически
полностью блокирует возможность
репликации или транскрипции ДНК. Ситуация
с 5-Br–dUrd, как соединением способным
инициировать транзиции, менее однозначна,
чем в случае 2-аминопурина. Дело в том,
что 5-Br-dUrd может образовывать кето- и
енольную формы. Минорная енольная форма
5-Br-Ura (рис. 5.12) возникает чаще, чем такая
же форма тимина, из-за большей
электроотрицательности Br по сравнению
CH3-группой
тимина.

|
Рис. 5.12 |
Высокая |
Поэтому,
более часто образуемая енольная форма
5-Br-Ura имеет тенденцию спариваться с
Gua, что приводит к транзиции A-T
G-C. в
норме же, кето-форма 5-Br-Ura, являющегося
аналогом тимина спаривается с Ade (рис.
5.13).
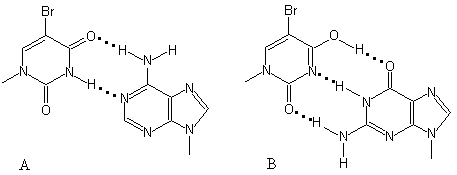
|
Рис. 5.13 |
В норме кето-форма |
Схема,
иллюстрирующая последовательность
этапов приводящих к транзиции A-T
G-C, вызванной енолизацией встроенного
в ДНК 5-Br-Ura показана на рис. 5.14.

|
Рис. 5.14 |
Схема появления |
Явление
интеркаляции.
Некоторые органические соединения,
которые характеризуются плоской
ароматической структурой с соответствующей
геометрией и размерами могут встраиваться
в ДНК между парами оснований —
интеркалировать.
В результате интеркаляции, эти соединения
вызывают появление вставок или делеций
одной или более пар оснований и тем
самым приводят к изменению рамки
считывания, если только вставки и
делеции не кратны трем парам оснований.
К таким интеркалирующим соединениям
относятся акридины
и этидий
бромид (рис.
5.15).

|
Рис. 5.15 |
Этидий бромид |
Рентгеноструктурный
анализ комплексов таких соединений с
синтетическими двухцепочечными
олигонуклеотидами показывает, что
плоские ароматические кольца акридиновых
красителей
внедряются между парами оснований
двойной спирали. Механизм внедрения
предполагает проникновение молекулы
красителя между парами оснований в
момент возникновения локального
нарушения структуры, при этом водородные
связи между парами оснований сохраняются,
тогда как «стэкинг»- взаимодействия
нарушаются.
Одним
из вариантов такого нарушения структуры
ДНК вследствие интеркаляции акридиновых
красителей
или
этидий
бромида
является образование изломов двойной
спирали в молекуле ДНК, которые получили
название кинков.
Химический
канцерогенез.
В настоящее время многие эксперты
считают, что в подавляющем большинстве
случаев заболевание раком инициируется
воздействием на нуклеиновые кислоты
определенных химических соединений.
Канцерогенные вещества поступают в
окружающую среду не только благодаря
синтезу и использованию новых химических
соединений в промышленных масштабах.
Канцерогенами являются также многие
соединения естественного происхождения.
Например, известными канцерогенами
являются афлатоксины,
продуцируемые некоторыми плесневыми
грибками. Несмотря на то, что интенсивное
изучение микотоксинов
началось сравнительно недавно, к
настоящему времени уже описано более
300 таких соединений, относящихся к 25
различным типам. Даже в небольших дозах
микотоксины оказывают разнообразные
токсические эффекты на человека и
животных, приводят к деградации печени,
геморрагии и карциноме. В качестве
главных по опасности микотоксинов
сейчас рассматривают группу метаболитов
гриба Aspergillus
flavus
– афлатоксинов, из которых наиболее
коварны афлатоксин
В1
и продукт его метаболического
гидроксилирования в организме коровы,
проникающий в молоко – афлатоксин
М1
(рис. 5.16). Доказано, что эти соединения
являются причиной цирроза и рака печени
у людей. Механизм действия афлатоксинов
состоит в том, что они после
15,16-эпоксидирования с участием печеночного
цитохрома Р-450 ковалентно связываются
с РНК, блокируя синтез белка.
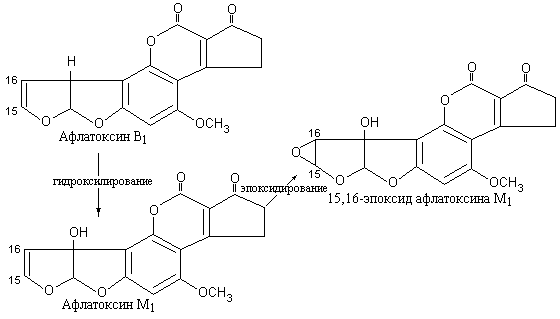
|
Рис. 5.16 |
Метаболическая |
К
другой группе канцерогенов, имеющих
естественное происхождение, относятся
такие соединения, как бензпирен
и бензантрацен,
являющиеся постоянными компонентами
табачного дыма, а также копченых
продуктов питания и продуктов
приготовляемых на углях. Хорошо известно,
что некоторые канцерогены характеризуются
непосредственным воздействием на
нуклеиновые кислоты, тогда как другие
(бензпирен и бензантрацен сами по себе
являются слабыми канцерогенами) прежде,
чем стать канцерогенными, должны пройти
стадию активации посредством
гидроксилирования и эпоксидирования
с участием монооксигеназных систем
печени. Ферменты, которые катализируют
активацию канцерогенов, принадлежат к
семейству цитохромов P-450. Как было
показано, конечным продуктом активации
бензпирена является вещество обладающее
мощным канцерогенным действием на
человека и животных и сильнейшим
мутагенным эффектом на бактериальные
клетки. Это соединение представляет
собой 7,8-дигидродиол-9,10-эпоксид
бензпирена.
Схема реакций, приводящих к метаболической
активации бензпирена приведена на рис.
5.17.
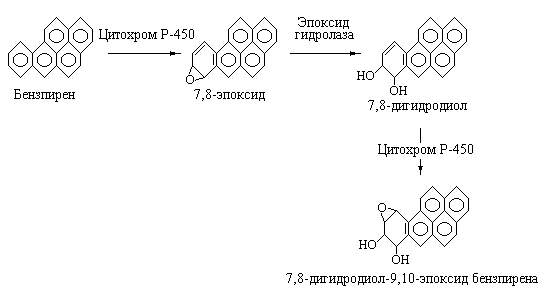
|
Рис. 5.17 |
Метаболическая |
На
первой стадии в описываемой
последовательности реакций бензпирен
под действием цитохрома Р-450 превращается
в 7,8-эпоксид, который далее с участием
эпоксидгидролазы присоединяет воду с
образованием 7,8-дигидродиола бензпирена.
На последней стадии, катализируемой
также цитохромом Р-450 образуется конечное
соединение 7,8-дигидродиол-9,10-эпоксид
бензпирена.
Следует
иметь в виду, что цитохромы P-450 обладают
уникальной способностью индуцироваться
неканцерогенными соединениями, такими
как этанол. Следовательно, алкоголь
может в значительной степени увеличивать
потенциальный риск рака в результате
воздействия канцерогенов.
Мутагенное
действие физических факторов
(X-лучи, УФ-,
-излучение).
Как ультрафиолетовое, так и рентгеновское
излучения являются сильнейшими
мутагенными средствами. Нормальные
основания, входящие в состав ДНК,
представлены, как известно, в виде кето-
и амино-форм,
находящихся в равновесии с очень
небольшими количествами минорных
енольной
и имино-форм.
Энергия УФ и X-лучей сдвигает это
равновесие в сторону образования
минорных таутомерных форм. В результате
чего повышенное содержание редких
таутомерных форм Ade и Cyt увеличивает
частоту их спаривания с Cyt и Ade,
соответственно (рис. 5.4 и 5.5). Считают,
что повышенное количество енольных
форм оснований в момент репликации
значительно повышает частоту мутаций
в новосинтезируемых цепях ДНК.
Действие
на ДНК жесткого излучения типа
рентгеновских лучей и -излучения
может приводить к изменению структуры
оснований. Результатом такого воздействия
может быть раскрытие гетероциклов,
разрушение фосфодиэфирных связей. В
присутствии кислорода накапливается
большое количество продуктов окисления
азотистых оснований и остатков
дезоксирибозы.
При
воздействии УФ-света, кроме сдвига
равновесия в сторону образования
минорных таутомерных форм оснований
происходит также появление, в первую
очередь, тиминовых димеров (рис. 5.18),
хотя в принципе возможно образование
ковалентно сшитых пар не только T-T, но
также T-C и C-C.

|
Рис. 5.18 |
Структура |
Соседние файлы в предмете [НЕСОРТИРОВАННОЕ]
- #
- #
- #
- #
- #
- #
- #
- #
- #
- #
- #
From Wikipedia, the free encyclopedia

Different types of indel mutation. Panel C is simply a deletion and not a frameshift mutation.
A frameshift mutation (also called a framing error or a reading frame shift) is a genetic mutation caused by indels (insertions or deletions) of a number of nucleotides in a DNA sequence that is not divisible by three. Due to the triplet nature of gene expression by codons, the insertion or deletion can change the reading frame (the grouping of the codons), resulting in a completely different translation from the original. The earlier in the sequence the deletion or insertion occurs, the more altered the protein.[1] A frameshift mutation is not the same as a single-nucleotide polymorphism in which a nucleotide is replaced, rather than inserted or deleted. A frameshift mutation will in general cause the reading of the codons after the mutation to code for different amino acids. The frameshift mutation will also alter the first stop codon («UAA», «UGA» or «UAG») encountered in the sequence. The polypeptide being created could be abnormally short or abnormally long, and will most likely not be functional.[2]
Frameshift mutations are apparent in severe genetic diseases such as Tay–Sachs disease; they increase susceptibility to certain cancers and classes of familial hypercholesterolaemia; in 1997,[3] a frameshift mutation was linked to resistance to infection by the HIV retrovirus. Frameshift mutations have been proposed as a source of biological novelty, as with the alleged creation of nylonase, however, this interpretation is controversial. A study by Negoro et al (2006)[4] found that a frameshift mutation was unlikely to have been the cause and that rather a two amino acid substitution in the active site of an ancestral esterase resulted in nylonase.
Background[edit]
The information contained in DNA determines protein function in the cells of all organisms. Transcription and translation allow this information to be communicated into making proteins. However, an error in reading this communication can cause protein function to be incorrect and eventually cause disease even as the cell incorporates a variety of corrective measures.
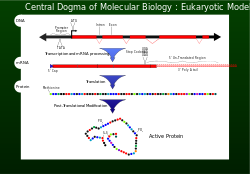
Central dogma[edit]
In 1956 Francis Crick described the flow of genetic information from DNA to a specific amino acid arrangement for making a protein as the central dogma.[1] For a cell to properly function, proteins are required to be produced accurately for structural and for catalytic activities. An incorrectly made protein can have detrimental effects on cell viability and in most cases cause the higher organism to become unhealthy by abnormal cellular functions. To ensure that the genome successfully passes the information on, proofreading mechanisms such as exonucleases and mismatch repair systems are incorporated in DNA replication .[1]
Transcription and translation[edit]
![]()
After DNA replication, the reading of a selected section of genetic information is accomplished by transcription.[1]
Nucleotides containing the genetic information are now on a single strand messenger template called mRNA. The mRNA is incorporated with a subunit of the ribosome and interacts with an rRNA. The genetic information carried in the codons of the mRNA are now read (decoded) by anticodons of the tRNA. As each codon (triplet) is read, amino acids are being joined together until a stop codon (UAG, UGA or UAA) is reached. At this point the polypeptide (protein) has been synthesised and is released.[1] For every 1000 amino acid incorporated into the protein, no more than one is incorrect. This fidelity of codon recognition, maintaining the importance of the proper reading frame, is accomplished by proper base pairing at the ribosome A site, GTP hydrolysis activity of EF-Tu a form of kinetic stability, and a proofreading mechanism as EF-Tu is released.[1]
Frameshifting may also occur during prophase translation, producing different proteins from overlapping open reading frames, such as the gag-pol-env retroviral proteins. This is fairly common in viruses and also occurs in bacteria and yeast (Farabaugh, 1996). Reverse transcriptase, as opposed to RNA Polymerase II, is thought to be a stronger cause of the occurrence of frameshift mutations. In experiments only 3–13% of all frameshift mutations occurred because of RNA Polymerase II. In prokaryotes the error rate inducing frameshift mutations is only somewhere in the range of .0001 and .00001.[5]
There are several biological processes that help to prevent frameshift mutations. Reverse mutations occur which change the mutated sequence back to the original wild type sequence. Another possibility for mutation correction is the use of a suppressor mutation. This offsets the effect of the original mutation by creating a secondary mutation, shifting the sequence to allow for the correct amino acids to be read. Guide RNA can also be used to insert or delete Uridine into the mRNA after transcription, this allows for the correct reading frame.[1]
Codon-triplet importance[edit]

The three letter code, the codon
A codon is a set of three nucleotides, a triplet that code for a certain amino acid. The first codon establishes the reading frame, whereby a new codon begins. A protein’s amino acid backbone sequence is defined by contiguous triplets.[6] Codons are key to translation of genetic information for the synthesis of proteins. The reading frame is set when translating the mRNA begins and is maintained as it reads one triplet to the next. The reading of the genetic code is subject to three rules the monitor codons in mRNA. First, codons are read in a 5′ to 3′ direction. Second, codons are nonoverlapping and the message has no gaps. The last rule, as stated above, that the message is translated in a fixed reading frame.[1]
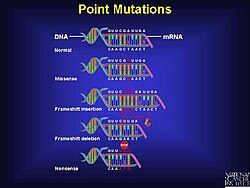
Example of different types of point mutations
Mechanism[edit]
Frameshift mutations can occur randomly or be caused by an external stimulus. The detection of frameshift mutations can occur via several different methods. Frameshifts are just one type of mutation that can lead to incomplete or incorrect proteins, but they account for a significant percentage of errors in DNA.
Genetic or environmental[edit]
This is a genetic mutation at the level of nucleotide bases. Why and how frameshift mutations occur are continually being sought after. An environmental study, specifically the production of UV-induced frameshift mutations by DNA polymerases deficient in 3′ → 5′ exonuclease activity was done. The normal sequence 5′ GTC GTT TTA CAA 3′ was changed to GTC GTT T TTA CAA (MIDT) of GTC GTT C TTA CAA (MIDC) to study frameshifts. E. coli pol I Kf and T7 DNA polymerase mutant enzymes devoid of 3′ → 5′ exonuclease activity produce UV-induced revertants at higher frequency than did their exonuclease proficient counterparts. The data indicates that loss of proofreading activity increases the frequency of UV-induced frameshifts.[7]
Detection[edit]
Fluorescence[edit]
The effects of neighboring bases and secondary structure to detect the frequency of frameshift mutations has been investigated in depth using fluorescence. Fluorescently tagged DNA, by means of base analogues, permits one to study the local changes of a DNA sequence.[8] Studies on the effects of the length of the primer strand reveal that an equilibrium mixture of four hybridization conformations was observed when template bases looped-out as a bulge, i.e. a structure flanked on both sides by duplex DNA. In contrast, a double-loop structure with an unusual unstacked DNA conformation at its downstream edge was observed when the extruded bases were positioned at the primer–template junction, showing that misalignments can be modified by neighboring DNA secondary structure.[9]
Sequencing[edit]
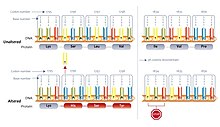
A deletion mutation alters every codon following it, and can make protein synthesis stop prematurely by forming a stop codon.
Sanger sequencing and pyrosequencing are two methods that have been used to detect frameshift mutations, however, it is likely that data generated will not be of the highest quality. Even still, 1.96 million indels have been identified through Sanger sequencing that do not overlap with other databases. When a frameshift mutation is observed it is compared against the Human Genome Mutation Database (HGMD) to determine if the mutation has a damaging effect. This is done by looking at four features. First, the ratio between the affected and conserved DNA, second the location of the mutation relative to the transcript, third the ratio of conserved and affected amino acids and finally the distance of the indel to the end of the exon.[10]
Massively Parallel Sequencing is a newer method that can be used to detect mutations. Using this method, up to 17 gigabases can be sequenced at once, as opposed to limited ranges for Sanger sequencing of only about 1 kilobase. Several technologies are available to perform this test and it is being looked at to be used in clinical applications.[11] When testing for different carcinomas, current methods only allow for looking at one gene at a time. Massively Parallel Sequencing can test for a variety of cancer causing mutations at once as opposed to several specific tests.[12] An experiment to determine the accuracy of this newer sequencing method tested for 21 genes and had no false positive calls for frameshift mutations.[13]
Diagnosis[edit]
A US patent (5,958,684) in 1999 by Leeuwen, details the methods and reagents for diagnosis of diseases caused by or associated with a gene having a somatic mutation giving rise to a frameshift mutation. The methods include providing a tissue or fluid sample and conducting gene analysis for frameshift mutation or a protein from this type of mutation. The nucleotide sequence of the suspected gene is provided from published gene sequences or from cloning and sequencing of the suspect gene. The amino acid sequence encoded by the gene is then predicted.[14]
Frequency[edit]
Despite the rules that govern the genetic code and the various mechanisms present in a cell to ensure the correct transfer of genetic information during the process of DNA replication as well as during translation, mutations do occur; frameshift mutation is not the only type. There are at least two other types of recognized point mutations, specifically missense mutation and nonsense mutation.[1] A frameshift mutation can drastically change the coding capacity (genetic information) of the message.[1] Small insertions or deletions (those less than 20 base pairs) make up 24% of mutations that manifest in currently recognized genetic disease.[10]
Frameshift mutations are found to be more common in repeat regions of DNA. A reason for this is because of slipping of the polymerase enzyme in repeat regions, allowing for mutations to enter the sequence.[15] Experiments can be run to determine the frequency of the frameshift mutation by adding or removing a pre-set number of nucleotides. Experiments have been run by adding four basepairs, called the +4 experiments, but a team from Emory University looked at the difference in frequency of the mutation by both adding and deleting a base pair. It was shown that there was no difference in the frequency between the addition and deletion of a base pair. There is however, a difference in the end result of the protein.[15]
Huntington’s disease is one of the nine codon reiteration disorders caused by polyglutamine expansion mutations that include spino-cerebellar ataxia (SCA) 1, 2, 6, 7 and 3, spinobulbar muscular atrophy and dentatorubal-pallidoluysianatrophy. There may be a link between diseases caused by polyglutamine and polyalanine expansion mutations, as frame shifting of the original SCA3 gene product encoding CAG/polyglutamines to GCA/polyalanines. Ribosomal slippage during translation of the SCA3 protein has been proposed as the mechanism resulting in shifting from the polyglutamine to the polyalanine-encoding frame. A dinucleotide deletion or single nucleotide insertion within the polyglutamine tract of huntingtin exon 1 would shift the CAG, polyglutamineen coding frame by +1 (+1 frame shift) to the GCA, polyalanine-encoding frame and introduce a novel epitope to the C terminus of Htt exon 1 (APAAAPAATRPGCG).[16]
Diseases[edit]
Several diseases have frameshift mutations as at least part of the cause. Knowing prevalent mutations can also aid in the diagnosis of the disease. Currently there are attempts to use frameshift mutations beneficially in the treatment of diseases, changing the reading frame of the amino acids.
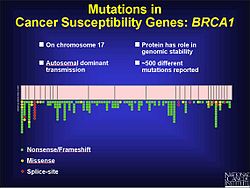
Frequency of mutations on BRCA1 gene on chromosome 17

Frequency of mutations on BRCA2 gene on chromosome 13
Cancer[edit]
Frameshift mutations are known to be a factor in colorectal cancer as well as other cancers with microsatellite instability. As stated previously, frameshift mutations are more likely to occur in a region of repeat sequence. When DNA mismatch repair does not fix the addition or deletion of bases, these mutations are more likely to be pathogenic. This may be in part because the tumor is not told to stop growing. Experiments in yeast and bacteria help to show characteristics of microsatellites that may contribute to defective DNA mismatch repair. These include the length of the microsatellite, the makeup of the genetic material and how pure the repeats are. Based on experimental results longer microsatellites have a higher rate of frameshift mutations. The flanking DNA can also contribute to frameshift mutations.[17] In prostate cancer a frameshift mutation changes the open reading frame (ORF) and prevents apoptosis from occurring. This leads to an unregulated growth of the tumor. While there are environmental factors that contribute to the progression of prostate cancer, there is also a genetic component. During testing of coding regions to identify mutations, 116 genetic variants were discovered, including 61 frameshift mutations.[18] There are over 500 mutations on chromosome 17 that seem to play a role in the development of breast and ovarian cancer in the BRCA1 gene, many of which are frameshift.[19]
Crohn’s disease[edit]
Crohn’s disease has an association with the NOD2 gene. The mutation is an insertion of a Cytosine at position 3020. This leads to a premature stop codon, shortening the protein that is supposed to be transcribed. When the protein is able to form normally, it responds to bacterial liposaccharides, where the 3020insC mutation prevents the protein from being responsive.[20]
Cystic fibrosis[edit]
Cystic fibrosis (CF) is a disease based on mutations in the CF transmembrane conductance regulator (CFTR) gene. There are over 1500 mutations identified, but not all cause the disease.[21] Most cases of cystic fibrosis are a result of the ∆F508 mutation, which deletes the entire amino acid. Two frameshift mutations are of interest in diagnosing CF, CF1213delT and CF1154-insTC. Both of these mutations commonly occur in tandem with at least one other mutation. They both lead to a small decrease in the function of the lungs and occur in about 1% of patients tested. These mutations were identified through Sanger sequencing.[22]
HIV[edit]
CCR5 is one of the cell entry co-factors associated with HIV, most frequently involved with nonsyncytium-inducing strains, is most apparent in HIV patients as opposed to AIDS patients. A 32 base pair deletion in CCR5 has been identified as a mutation that negates the likelihood of an HIV infection. This region on the open reading frame ORF contains a frameshift mutation leading to a premature stop codon. This leads to the loss of the HIV-coreceptor function in vitro. CCR5-1 is considered the wild type and CCR5-2 is considered to be the mutant allele. Those with a heterozygous mutation for the CCR5 were less susceptible to the development of HIV. In a study, despite high exposure to the HIV virus, there was no one homozygous for the CCR5 mutation that tested positive for HIV.[3]
Tay–Sachs disease[edit]
Tay–Sachs disease is a fatal disease affecting the central nervous system. It is most frequently found in infants and small children. Disease progression begins in the womb but symptoms do not appear until approximately 6 months of age. There is no cure for the disease.[23] Mutations in the β-hexosaminidase A (Hex A) gene are known to affect the onset of Tay-Sachs, with 78 mutations of different types being described, 67 of which are known to cause disease. Most of the mutations observed (65/78) are single base substitutions or SNPs, 11 deletions, 1 large and 10 small, and 2 insertions. 8 of the observed mutations are frameshift, 6 deletions and 2 insertions. A 4 base pair insertion in exon 11 is observed in 80% of Tay-Sachs disease presence in the Ashkenazi Jewish population. The frameshift mutations lead to an early stop codon which is known to play a role in the disease in infants. Delayed onset disease appears to be caused by 4 different mutations, one being a 3 base pair deletion.[24]
Smith–Magenis syndrome[edit]
Smith–Magenis syndrome (SMS) is a complex syndrome involving intellectual disabilities, sleep disturbance, behavioural problems, and a variety of craniofacial, skeletal, and visceral anomalies. The majority of SMS cases harbor an ~3.5 Mb common deletion that encompasses the retinoic acid induced-1 (RAI1) gene. Other cases illustrate variability in the SMS phenotype not previously shown for RAI1 mutation, including hearing loss, absence of self-abusive behaviours, and mild global delays. Sequencing of RAI1 revealed mutation of a heptamericC-tract (CCCCCCC) in exon 3 resulting in frameshift mutations. Of the seven reported frameshift mutations occurring in poly C-tracts in RAI1, four cases (~57%) occur at this heptameric C-tract. The results indicate that this heptameric C-tract is a preferential recombination hotspot insertion/deletions (SNindels) and therefore a primary target for analysis in patients suspected for mutations in RAI1.[25]
Hypertrophic cardiomyopathy[edit]
Hypertrophic cardiomyopathy is the most common cause of sudden death in young people, including trained athletes, and is caused by mutations in genes encoding proteins of the cardiac sarcomere. Mutations in the Troponin C gene (TNNC1) are a rare genetic cause of hypertrophic cardiomyopathy. A recent study has indicated that a frameshift mutation (c.363dupG or p.Gln122AlafsX30) in Troponin C was the cause of hypertrophic cardiomyopathy (and sudden cardiac death) in a 19-year-old male.[26]
Cures[edit]
Finding a cure for the diseases caused by frameshift mutations is rare. Research into this is ongoing. One example is a primary immunodeficiency (PID), an inherited condition which can lead to an increase in infections. There are 120 genes and 150 mutations that play a role in primary immunodeficiencies. The standard treatment is currently gene therapy, but this is a highly risky treatment and can often lead to other diseases, such as leukemia. Gene therapy procedures include modifying the zinc fringer nuclease fusion protein, cleaving both ends of the mutation, which in turn removes it from the sequence. Antisense-oligonucleotide mediated exon skipping is another possibility for Duchenne muscular dystrophy. This process allows for passing over the mutation so that the rest of the sequence remains in frame and the function of the protein stays intact. This, however, does not cure the disease, just treats symptoms, and is only practical in structural proteins or other repetitive genes. A third form of repair is revertant mosaicism, which is naturally occurring by creating a reverse mutation or a mutation at a second site that corrects the reading frame. This reversion may happen by intragenic recombination, mitotic gene conversion, second site DNA slipping or site-specific reversion. This is possible in several diseases, such as X-linked severe combined immunodeficiency (SCID), Wiskott–Aldrich syndrome, and Bloom syndrome. There are no drugs or other pharmacogenomic methods that help with PIDs.[27]
A European patent (EP1369126A1) in 2003 by Bork records a method used for prevention of cancers and for the curative treatment of cancers and precancers such as DNA-mismatch repair deficient (MMR) sporadic tumours and HNPCC associated tumours. The idea is to use immunotherapy with combinatorial mixtures of tumour-specific frameshift mutation-derived peptides to elicit a cytotoxic T-cell response specifically directed against tumour cells.[28]
See also[edit]
- Translational frameshift
- Mutation
- Transcription (genetics)
- Translation (biology)
- codon
- protein
- reading frame
- point mutation
- Crohn’s disease
- Tay–Sachs disease
References[edit]
- ^ a b c d e f g h i j Losick, Richard; Watson, James D.; Baker, Tania A.; Bell, Stephen; Gann, Alexander; Levine, Michael W. (2008). Molecular biology of the gene (6th ed.). San Francisco: Pearson/Benjamin Cummings. ISBN 978-0-8053-9592-1.
- ^ «DNA Is Constantly Changing through the Process of Mutation». Nature. Retrieved 17 May 2019.
- ^ a b Zimmerman PA, Buckler-White A, Alkhatib G, Spalding T, Kubofcik J, Combadiere C, Weissman D, Cohen O, Rubbert A, Lam G, Vaccarezza M, Kennedy PE, Kumaraswami V, Giorgi JV, Detels R, Hunter J, Chopek M, Berger EA, Fauci AS, Nutman TB, Murphy PM (January 1997). «Inherited resistance to HIV-1 conferred by an inactivating mutation in CC chemokine receptor 5: studies in populations with contrasting clinical phenotypes, defined racial background, and quantified risk». Molecular Medicine (Cambridge, Mass.). 3 (1): 23–36. PMC 2230106. PMID 9132277.
- ^ Negoro S, Ohki T, Shibata N, Mizuno N, Wakitani Y, Tsurukame J, Matsumoto K, Kawamoto I, Takeo M, Higuchi Y (November 2005). «X-ray crystallographic analysis of 6-aminohexanoate-dimer hydrolase: molecular basis for the birth of a nylon oligomer-degrading enzyme». J Biol Chem. 280 (47): 39644–52. doi:10.1074/jbc.m505946200. PMID 16162506.
- ^ Zhang, J (August 2004). «Host RNA polymerase II makes minimal contributions to retroviral frame-shift mutations». The Journal of General Virology. 85 (Pt 8): 2389–95. doi:10.1099/vir.0.80081-0. PMID 15269381.
- ^ Cox, Michael; Nelson, David R.; Lehninger, Albert L (2008). Lehninger principles of biochemistry. San Francisco: W.H. Freeman. ISBN 978-0-7167-7108-1.
- ^ Sagher, Daphna; Turkington, Edith; Acharya, Sonia; Strauss, Bernard (July 1994). «Production of UV-induced Frameshift Mutations in Vitro by DNA Polymerases Deficient in 3′ → 5′ Exonuclease Activity». Journal of Molecular Biology. 240 (3): 226–242. doi:10.1006/jmbi.1994.1437. PMID 8028006.
- ^ Johnson, Neil P.; Walter A. Baase; Peter H. von Hippel (March 2004). «Low-energy circular dichroism of 2-aminopurine dinucleotide as a probe of local conformation of DNA and RNA». Proc Natl Acad Sci U S A. 101 (10): 3426–31. Bibcode:2004PNAS..101.3426J. doi:10.1073/pnas.0400591101. PMC 373478. PMID 14993592.
- ^ Baase, Walter A.; Davis Jose; Benjamin C. Ponedel; Peter H. von Hippel; Neil P. Johnson (2009). «DNA models of trinucleotide frameshift deletions: the formation of loops and bulges at the primer–template junction». Nucleic Acids Research. 37 (5): 1682–9. doi:10.1093/nar/gkn1042. PMC 2655659. PMID 19155277.
- ^ a b Hu, J; Ng, PC (9 February 2012). «Predicting the effects of frameshifting indels». Genome Biology. 13 (2): R9. doi:10.1186/gb-2012-13-2-r9. PMC 3334572. PMID 22322200.
- ^ Tucker, Tracy; Marra, Marco; Friedman, Jan M. (2009). «Massively Parallel Sequencing: The Next Big Thing in Genetic Medicine». The American Journal of Human Genetics. 85 (2): 142–154. doi:10.1016/j.ajhg.2009.06.022. PMC 2725244. PMID 19679224.
- ^ Walsh, T.; Casadei, S.; Lee, M. K.; Pennil, C. C.; Nord, A. S.; Thornton, A. M.; Roeb, W.; Agnew, K. J.; Stray, S. M.; Wickramanayake, A.; Norquist, B.; Pennington, K. P.; Garcia, R. L.; King, M.-C.; Swisher, E. M. (2011). «From the Cover: Mutations in 12 genes for inherited ovarian, fallopian tube, and peritoneal carcinoma identified by massively parallel sequencing». Proc Natl Acad Sci U S A. 108 (44): 18032–7. Bibcode:2011PNAS..10818032W. doi:10.1073/pnas.1115052108. PMC 3207658. PMID 22006311.
- ^ Walsh, T.; Lee, M. K.; Casadei, S.; Thornton, A. M.; Stray, S. M.; Pennil, C.; Nord, A. S.; Mandell, J. B.; Swisher, E. M.; King, M.-C. (2010). «Detection of inherited mutations for breast and ovarian cancer using genomic capture and massively parallel sequencing». Proc Natl Acad Sci U S A. 107 (28): 12629–33. Bibcode:2010PNAS..10712629W. doi:10.1073/pnas.1007983107. PMC 2906584. PMID 20616022.
- ^ US Patent 5,958,684 (September 28, 1999) «Diagnosis of Neurodegenerative Disease» by Leeuwen et al
- ^ a b Harfe, BD; Jinks-Robertson, S (July 1999). «Removal of frameshift intermediates by mismatch repair proteins in Saccharomyces cerevisiae». Molecular and Cellular Biology. 19 (7): 4766–73. doi:10.1128/MCB.19.7.4766. PMC 84275. PMID 10373526.
- ^ Davies, J E; Rubinsztein, D C (2006). «Polyalanine and polyserine frameshift products in Huntington’s disease». Journal of Medical Genetics. 43 (11): 893–896. doi:10.1136/jmg.2006.044222. PMC 2563184. PMID 16801344.
- ^ Schmoldt, A; Benthe, HF; Haberland, G (1 September 1975). «Digitoxin metabolism by rat liver microsomes». Biochemical Pharmacology. 24 (17): 1639–41. doi:10.1016/0006-2952(75)90094-5. PMID 10.
- ^ Xu, XiaoLin; Zhu, KaiChang; Liu, Feng; Wang, Yue; Shen, JianGuo; Jin, Jizhong; Wang, Zhong; Chen, Lin; Li, Jiadong; Xu, Min (May 2013). «Identification of somatic mutations in human prostate cancer by RNA-Seq». Gene. 519 (2): 343–7. doi:10.1016/j.gene.2013.01.046. PMID 23434521.
- ^ «Cancer Genomics». National Cancer Institute at the National Institute of Health. Archived from the original on 18 March 2013. Retrieved 24 March 2013.
- ^ Ogura Y, Bonen DK, Inohara N, Nicolae DL, Chen FF, Ramos R, Britton H, Moran T, Karaliuskas R, Duerr RH, Achkar JP, Brant SR, Bayless TM, Kirschner BS, Hanauer SB, Nuñez G, Cho JH (May 31, 2001). «A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease» (PDF). Nature. 411 (6837): 603–6. Bibcode:2001Natur.411..603O. doi:10.1038/35079114. hdl:2027.42/62856. PMID 11385577. S2CID 205017657.
- ^ Farrell PM, Rosenstein BJ, White TB, Accurso FJ, Castellani C, Cutting GR, Durie PR, Legrys VA, Massie J, Parad RB, Rock MJ, Campbell PW (2008). «Guidelines for Diagnosis of Cystic Fibrosis in Newborns through Older Adults: Cystic Fibrosis Foundation Consensus Report». The Journal of Pediatrics. 153 (2): S4–S14. doi:10.1016/j.jpeds.2008.05.005. PMC 2810958. PMID 18639722.
- ^ Iannuzzi, MC; Stern, RC; Collins, FS; Hon, CT; Hidaka, N; Strong, T; Becker, L; Drumm, ML; White, MB; Gerrard, B (February 1991). «Two frameshift mutations in the cystic fibrosis gene». American Journal of Human Genetics. 48 (2): 227–31. PMC 1683026. PMID 1990834.
- ^ «Learning About Tay-Sachs Disease». National Human Genome Research Institute. Retrieved 24 March 2013.
- ^ Myerowitz, R (1997). «Tay-Sachs disease-causing mutations and neutral polymorphisms in the Hex A gene». Human Mutation. 9 (3): 195–208. doi:10.1002/(SICI)1098-1004(1997)9:3<195::AID-HUMU1>3.0.CO;2-7. PMID 9090523.
- ^ Truong, Hoa T; Dudding, Tracy; Blanchard, Christopher L.; Elsea, Sarah H (2010). «Frameshift mutation hotspot identified in Smith-Magenis syndrome: case report and review of literature». BMC Medical Genetics. 11 (1): 142. doi:10.1186/1471-2350-11-142. PMC 2964533. PMID 20932317.
- ^ Chung WK, Kitner C, Maron BJ (June 2011). «Novel frameshift mutation in Troponin C ( TNNC1) associated with hypertrophic cardiomyopathy and sudden death». Cardiol Young. 21 (3): 345–8. doi:10.1017/S1047951110001927. PMID 21262074. S2CID 46682245.
- ^ Hu, Hailiang; Gatti, Richard A (2008). «New approaches to treatment of primary immunodeficiencies: fixing mutations with chemicals». Current Opinion in Allergy and Clinical Immunology. 8 (6): 540–6. doi:10.1097/ACI.0b013e328314b63b. PMC 2686128. PMID 18978469.
- ^ European Patent [1] (December 10, 2003) «Use of coding microsatellite region frameshift mutation-derived peptides for treating cancer» by Bork et al
Further reading[edit]
- Farabaugh PJ (1996). «Programmed translational frameshifting». Annu. Rev. Genet. 30 (1): 507–28. doi:10.1146/annurev.genet.30.1.507. PMC 239420. PMID 8982463.
- Lewis, Ricki (2005). Human Genetics: Concepts and Applications (6th ed.). Boston MA: McGraw Hill. pp. 227–8. ISBN 978-0-07-111156-0.
- «Nylonase Enzymes». 20 April 2004. Retrieved 2 June 2009.
External links[edit]
- Frameshift+Mutation at the US National Library of Medicine Medical Subject Headings (MeSH)
- NCBI dbSNP database — «a central repository for both single base nucleotide substitutions and short deletion and insertion polymorphisms»
- Wise2 — aligns a protein against a DNA sequence allowing frameshifts and introns
- FastY — compare a DNA sequence to a protein sequence database, allowing gaps and frameshifts
- Path — tool that compares two frameshift proteins (back-translation principle)
- HGMD — Human Genome Mutation Database
From Wikipedia, the free encyclopedia
Different types of indel mutation. Panel C is simply a deletion and not a frameshift mutation.
A frameshift mutation (also called a framing error or a reading frame shift) is a genetic mutation caused by indels (insertions or deletions) of a number of nucleotides in a DNA sequence that is not divisible by three. Due to the triplet nature of gene expression by codons, the insertion or deletion can change the reading frame (the grouping of the codons), resulting in a completely different translation from the original. The earlier in the sequence the deletion or insertion occurs, the more altered the protein.[1] A frameshift mutation is not the same as a single-nucleotide polymorphism in which a nucleotide is replaced, rather than inserted or deleted. A frameshift mutation will in general cause the reading of the codons after the mutation to code for different amino acids. The frameshift mutation will also alter the first stop codon («UAA», «UGA» or «UAG») encountered in the sequence. The polypeptide being created could be abnormally short or abnormally long, and will most likely not be functional.[2]
Frameshift mutations are apparent in severe genetic diseases such as Tay–Sachs disease; they increase susceptibility to certain cancers and classes of familial hypercholesterolaemia; in 1997,[3] a frameshift mutation was linked to resistance to infection by the HIV retrovirus. Frameshift mutations have been proposed as a source of biological novelty, as with the alleged creation of nylonase, however, this interpretation is controversial. A study by Negoro et al (2006)[4] found that a frameshift mutation was unlikely to have been the cause and that rather a two amino acid substitution in the active site of an ancestral esterase resulted in nylonase.
Background[edit]
The information contained in DNA determines protein function in the cells of all organisms. Transcription and translation allow this information to be communicated into making proteins. However, an error in reading this communication can cause protein function to be incorrect and eventually cause disease even as the cell incorporates a variety of corrective measures.
Central dogma[edit]
In 1956 Francis Crick described the flow of genetic information from DNA to a specific amino acid arrangement for making a protein as the central dogma.[1] For a cell to properly function, proteins are required to be produced accurately for structural and for catalytic activities. An incorrectly made protein can have detrimental effects on cell viability and in most cases cause the higher organism to become unhealthy by abnormal cellular functions. To ensure that the genome successfully passes the information on, proofreading mechanisms such as exonucleases and mismatch repair systems are incorporated in DNA replication .[1]
Transcription and translation[edit]
![]()
After DNA replication, the reading of a selected section of genetic information is accomplished by transcription.[1]
Nucleotides containing the genetic information are now on a single strand messenger template called mRNA. The mRNA is incorporated with a subunit of the ribosome and interacts with an rRNA. The genetic information carried in the codons of the mRNA are now read (decoded) by anticodons of the tRNA. As each codon (triplet) is read, amino acids are being joined together until a stop codon (UAG, UGA or UAA) is reached. At this point the polypeptide (protein) has been synthesised and is released.[1] For every 1000 amino acid incorporated into the protein, no more than one is incorrect. This fidelity of codon recognition, maintaining the importance of the proper reading frame, is accomplished by proper base pairing at the ribosome A site, GTP hydrolysis activity of EF-Tu a form of kinetic stability, and a proofreading mechanism as EF-Tu is released.[1]
Frameshifting may also occur during prophase translation, producing different proteins from overlapping open reading frames, such as the gag-pol-env retroviral proteins. This is fairly common in viruses and also occurs in bacteria and yeast (Farabaugh, 1996). Reverse transcriptase, as opposed to RNA Polymerase II, is thought to be a stronger cause of the occurrence of frameshift mutations. In experiments only 3–13% of all frameshift mutations occurred because of RNA Polymerase II. In prokaryotes the error rate inducing frameshift mutations is only somewhere in the range of .0001 and .00001.[5]
There are several biological processes that help to prevent frameshift mutations. Reverse mutations occur which change the mutated sequence back to the original wild type sequence. Another possibility for mutation correction is the use of a suppressor mutation. This offsets the effect of the original mutation by creating a secondary mutation, shifting the sequence to allow for the correct amino acids to be read. Guide RNA can also be used to insert or delete Uridine into the mRNA after transcription, this allows for the correct reading frame.[1]
Codon-triplet importance[edit]
The three letter code, the codon
A codon is a set of three nucleotides, a triplet that code for a certain amino acid. The first codon establishes the reading frame, whereby a new codon begins. A protein’s amino acid backbone sequence is defined by contiguous triplets.[6] Codons are key to translation of genetic information for the synthesis of proteins. The reading frame is set when translating the mRNA begins and is maintained as it reads one triplet to the next. The reading of the genetic code is subject to three rules the monitor codons in mRNA. First, codons are read in a 5′ to 3′ direction. Second, codons are nonoverlapping and the message has no gaps. The last rule, as stated above, that the message is translated in a fixed reading frame.[1]
Example of different types of point mutations
Mechanism[edit]
Frameshift mutations can occur randomly or be caused by an external stimulus. The detection of frameshift mutations can occur via several different methods. Frameshifts are just one type of mutation that can lead to incomplete or incorrect proteins, but they account for a significant percentage of errors in DNA.
Genetic or environmental[edit]
This is a genetic mutation at the level of nucleotide bases. Why and how frameshift mutations occur are continually being sought after. An environmental study, specifically the production of UV-induced frameshift mutations by DNA polymerases deficient in 3′ → 5′ exonuclease activity was done. The normal sequence 5′ GTC GTT TTA CAA 3′ was changed to GTC GTT T TTA CAA (MIDT) of GTC GTT C TTA CAA (MIDC) to study frameshifts. E. coli pol I Kf and T7 DNA polymerase mutant enzymes devoid of 3′ → 5′ exonuclease activity produce UV-induced revertants at higher frequency than did their exonuclease proficient counterparts. The data indicates that loss of proofreading activity increases the frequency of UV-induced frameshifts.[7]
Detection[edit]
Fluorescence[edit]
The effects of neighboring bases and secondary structure to detect the frequency of frameshift mutations has been investigated in depth using fluorescence. Fluorescently tagged DNA, by means of base analogues, permits one to study the local changes of a DNA sequence.[8] Studies on the effects of the length of the primer strand reveal that an equilibrium mixture of four hybridization conformations was observed when template bases looped-out as a bulge, i.e. a structure flanked on both sides by duplex DNA. In contrast, a double-loop structure with an unusual unstacked DNA conformation at its downstream edge was observed when the extruded bases were positioned at the primer–template junction, showing that misalignments can be modified by neighboring DNA secondary structure.[9]
Sequencing[edit]
A deletion mutation alters every codon following it, and can make protein synthesis stop prematurely by forming a stop codon.
Sanger sequencing and pyrosequencing are two methods that have been used to detect frameshift mutations, however, it is likely that data generated will not be of the highest quality. Even still, 1.96 million indels have been identified through Sanger sequencing that do not overlap with other databases. When a frameshift mutation is observed it is compared against the Human Genome Mutation Database (HGMD) to determine if the mutation has a damaging effect. This is done by looking at four features. First, the ratio between the affected and conserved DNA, second the location of the mutation relative to the transcript, third the ratio of conserved and affected amino acids and finally the distance of the indel to the end of the exon.[10]
Massively Parallel Sequencing is a newer method that can be used to detect mutations. Using this method, up to 17 gigabases can be sequenced at once, as opposed to limited ranges for Sanger sequencing of only about 1 kilobase. Several technologies are available to perform this test and it is being looked at to be used in clinical applications.[11] When testing for different carcinomas, current methods only allow for looking at one gene at a time. Massively Parallel Sequencing can test for a variety of cancer causing mutations at once as opposed to several specific tests.[12] An experiment to determine the accuracy of this newer sequencing method tested for 21 genes and had no false positive calls for frameshift mutations.[13]
Diagnosis[edit]
A US patent (5,958,684) in 1999 by Leeuwen, details the methods and reagents for diagnosis of diseases caused by or associated with a gene having a somatic mutation giving rise to a frameshift mutation. The methods include providing a tissue or fluid sample and conducting gene analysis for frameshift mutation or a protein from this type of mutation. The nucleotide sequence of the suspected gene is provided from published gene sequences or from cloning and sequencing of the suspect gene. The amino acid sequence encoded by the gene is then predicted.[14]
Frequency[edit]
Despite the rules that govern the genetic code and the various mechanisms present in a cell to ensure the correct transfer of genetic information during the process of DNA replication as well as during translation, mutations do occur; frameshift mutation is not the only type. There are at least two other types of recognized point mutations, specifically missense mutation and nonsense mutation.[1] A frameshift mutation can drastically change the coding capacity (genetic information) of the message.[1] Small insertions or deletions (those less than 20 base pairs) make up 24% of mutations that manifest in currently recognized genetic disease.[10]
Frameshift mutations are found to be more common in repeat regions of DNA. A reason for this is because of slipping of the polymerase enzyme in repeat regions, allowing for mutations to enter the sequence.[15] Experiments can be run to determine the frequency of the frameshift mutation by adding or removing a pre-set number of nucleotides. Experiments have been run by adding four basepairs, called the +4 experiments, but a team from Emory University looked at the difference in frequency of the mutation by both adding and deleting a base pair. It was shown that there was no difference in the frequency between the addition and deletion of a base pair. There is however, a difference in the end result of the protein.[15]
Huntington’s disease is one of the nine codon reiteration disorders caused by polyglutamine expansion mutations that include spino-cerebellar ataxia (SCA) 1, 2, 6, 7 and 3, spinobulbar muscular atrophy and dentatorubal-pallidoluysianatrophy. There may be a link between diseases caused by polyglutamine and polyalanine expansion mutations, as frame shifting of the original SCA3 gene product encoding CAG/polyglutamines to GCA/polyalanines. Ribosomal slippage during translation of the SCA3 protein has been proposed as the mechanism resulting in shifting from the polyglutamine to the polyalanine-encoding frame. A dinucleotide deletion or single nucleotide insertion within the polyglutamine tract of huntingtin exon 1 would shift the CAG, polyglutamineen coding frame by +1 (+1 frame shift) to the GCA, polyalanine-encoding frame and introduce a novel epitope to the C terminus of Htt exon 1 (APAAAPAATRPGCG).[16]
Diseases[edit]
Several diseases have frameshift mutations as at least part of the cause. Knowing prevalent mutations can also aid in the diagnosis of the disease. Currently there are attempts to use frameshift mutations beneficially in the treatment of diseases, changing the reading frame of the amino acids.
Frequency of mutations on BRCA1 gene on chromosome 17
Frequency of mutations on BRCA2 gene on chromosome 13
Cancer[edit]
Frameshift mutations are known to be a factor in colorectal cancer as well as other cancers with microsatellite instability. As stated previously, frameshift mutations are more likely to occur in a region of repeat sequence. When DNA mismatch repair does not fix the addition or deletion of bases, these mutations are more likely to be pathogenic. This may be in part because the tumor is not told to stop growing. Experiments in yeast and bacteria help to show characteristics of microsatellites that may contribute to defective DNA mismatch repair. These include the length of the microsatellite, the makeup of the genetic material and how pure the repeats are. Based on experimental results longer microsatellites have a higher rate of frameshift mutations. The flanking DNA can also contribute to frameshift mutations.[17] In prostate cancer a frameshift mutation changes the open reading frame (ORF) and prevents apoptosis from occurring. This leads to an unregulated growth of the tumor. While there are environmental factors that contribute to the progression of prostate cancer, there is also a genetic component. During testing of coding regions to identify mutations, 116 genetic variants were discovered, including 61 frameshift mutations.[18] There are over 500 mutations on chromosome 17 that seem to play a role in the development of breast and ovarian cancer in the BRCA1 gene, many of which are frameshift.[19]
Crohn’s disease[edit]
Crohn’s disease has an association with the NOD2 gene. The mutation is an insertion of a Cytosine at position 3020. This leads to a premature stop codon, shortening the protein that is supposed to be transcribed. When the protein is able to form normally, it responds to bacterial liposaccharides, where the 3020insC mutation prevents the protein from being responsive.[20]
Cystic fibrosis[edit]
Cystic fibrosis (CF) is a disease based on mutations in the CF transmembrane conductance regulator (CFTR) gene. There are over 1500 mutations identified, but not all cause the disease.[21] Most cases of cystic fibrosis are a result of the ∆F508 mutation, which deletes the entire amino acid. Two frameshift mutations are of interest in diagnosing CF, CF1213delT and CF1154-insTC. Both of these mutations commonly occur in tandem with at least one other mutation. They both lead to a small decrease in the function of the lungs and occur in about 1% of patients tested. These mutations were identified through Sanger sequencing.[22]
HIV[edit]
CCR5 is one of the cell entry co-factors associated with HIV, most frequently involved with nonsyncytium-inducing strains, is most apparent in HIV patients as opposed to AIDS patients. A 32 base pair deletion in CCR5 has been identified as a mutation that negates the likelihood of an HIV infection. This region on the open reading frame ORF contains a frameshift mutation leading to a premature stop codon. This leads to the loss of the HIV-coreceptor function in vitro. CCR5-1 is considered the wild type and CCR5-2 is considered to be the mutant allele. Those with a heterozygous mutation for the CCR5 were less susceptible to the development of HIV. In a study, despite high exposure to the HIV virus, there was no one homozygous for the CCR5 mutation that tested positive for HIV.[3]
Tay–Sachs disease[edit]
Tay–Sachs disease is a fatal disease affecting the central nervous system. It is most frequently found in infants and small children. Disease progression begins in the womb but symptoms do not appear until approximately 6 months of age. There is no cure for the disease.[23] Mutations in the β-hexosaminidase A (Hex A) gene are known to affect the onset of Tay-Sachs, with 78 mutations of different types being described, 67 of which are known to cause disease. Most of the mutations observed (65/78) are single base substitutions or SNPs, 11 deletions, 1 large and 10 small, and 2 insertions. 8 of the observed mutations are frameshift, 6 deletions and 2 insertions. A 4 base pair insertion in exon 11 is observed in 80% of Tay-Sachs disease presence in the Ashkenazi Jewish population. The frameshift mutations lead to an early stop codon which is known to play a role in the disease in infants. Delayed onset disease appears to be caused by 4 different mutations, one being a 3 base pair deletion.[24]
Smith–Magenis syndrome[edit]
Smith–Magenis syndrome (SMS) is a complex syndrome involving intellectual disabilities, sleep disturbance, behavioural problems, and a variety of craniofacial, skeletal, and visceral anomalies. The majority of SMS cases harbor an ~3.5 Mb common deletion that encompasses the retinoic acid induced-1 (RAI1) gene. Other cases illustrate variability in the SMS phenotype not previously shown for RAI1 mutation, including hearing loss, absence of self-abusive behaviours, and mild global delays. Sequencing of RAI1 revealed mutation of a heptamericC-tract (CCCCCCC) in exon 3 resulting in frameshift mutations. Of the seven reported frameshift mutations occurring in poly C-tracts in RAI1, four cases (~57%) occur at this heptameric C-tract. The results indicate that this heptameric C-tract is a preferential recombination hotspot insertion/deletions (SNindels) and therefore a primary target for analysis in patients suspected for mutations in RAI1.[25]
Hypertrophic cardiomyopathy[edit]
Hypertrophic cardiomyopathy is the most common cause of sudden death in young people, including trained athletes, and is caused by mutations in genes encoding proteins of the cardiac sarcomere. Mutations in the Troponin C gene (TNNC1) are a rare genetic cause of hypertrophic cardiomyopathy. A recent study has indicated that a frameshift mutation (c.363dupG or p.Gln122AlafsX30) in Troponin C was the cause of hypertrophic cardiomyopathy (and sudden cardiac death) in a 19-year-old male.[26]
Cures[edit]
Finding a cure for the diseases caused by frameshift mutations is rare. Research into this is ongoing. One example is a primary immunodeficiency (PID), an inherited condition which can lead to an increase in infections. There are 120 genes and 150 mutations that play a role in primary immunodeficiencies. The standard treatment is currently gene therapy, but this is a highly risky treatment and can often lead to other diseases, such as leukemia. Gene therapy procedures include modifying the zinc fringer nuclease fusion protein, cleaving both ends of the mutation, which in turn removes it from the sequence. Antisense-oligonucleotide mediated exon skipping is another possibility for Duchenne muscular dystrophy. This process allows for passing over the mutation so that the rest of the sequence remains in frame and the function of the protein stays intact. This, however, does not cure the disease, just treats symptoms, and is only practical in structural proteins or other repetitive genes. A third form of repair is revertant mosaicism, which is naturally occurring by creating a reverse mutation or a mutation at a second site that corrects the reading frame. This reversion may happen by intragenic recombination, mitotic gene conversion, second site DNA slipping or site-specific reversion. This is possible in several diseases, such as X-linked severe combined immunodeficiency (SCID), Wiskott–Aldrich syndrome, and Bloom syndrome. There are no drugs or other pharmacogenomic methods that help with PIDs.[27]
A European patent (EP1369126A1) in 2003 by Bork records a method used for prevention of cancers and for the curative treatment of cancers and precancers such as DNA-mismatch repair deficient (MMR) sporadic tumours and HNPCC associated tumours. The idea is to use immunotherapy with combinatorial mixtures of tumour-specific frameshift mutation-derived peptides to elicit a cytotoxic T-cell response specifically directed against tumour cells.[28]
See also[edit]
- Translational frameshift
- Mutation
- Transcription (genetics)
- Translation (biology)
- codon
- protein
- reading frame
- point mutation
- Crohn’s disease
- Tay–Sachs disease
References[edit]
- ^ a b c d e f g h i j Losick, Richard; Watson, James D.; Baker, Tania A.; Bell, Stephen; Gann, Alexander; Levine, Michael W. (2008). Molecular biology of the gene (6th ed.). San Francisco: Pearson/Benjamin Cummings. ISBN 978-0-8053-9592-1.
- ^ «DNA Is Constantly Changing through the Process of Mutation». Nature. Retrieved 17 May 2019.
- ^ a b Zimmerman PA, Buckler-White A, Alkhatib G, Spalding T, Kubofcik J, Combadiere C, Weissman D, Cohen O, Rubbert A, Lam G, Vaccarezza M, Kennedy PE, Kumaraswami V, Giorgi JV, Detels R, Hunter J, Chopek M, Berger EA, Fauci AS, Nutman TB, Murphy PM (January 1997). «Inherited resistance to HIV-1 conferred by an inactivating mutation in CC chemokine receptor 5: studies in populations with contrasting clinical phenotypes, defined racial background, and quantified risk». Molecular Medicine (Cambridge, Mass.). 3 (1): 23–36. PMC 2230106. PMID 9132277.
- ^ Negoro S, Ohki T, Shibata N, Mizuno N, Wakitani Y, Tsurukame J, Matsumoto K, Kawamoto I, Takeo M, Higuchi Y (November 2005). «X-ray crystallographic analysis of 6-aminohexanoate-dimer hydrolase: molecular basis for the birth of a nylon oligomer-degrading enzyme». J Biol Chem. 280 (47): 39644–52. doi:10.1074/jbc.m505946200. PMID 16162506.
- ^ Zhang, J (August 2004). «Host RNA polymerase II makes minimal contributions to retroviral frame-shift mutations». The Journal of General Virology. 85 (Pt 8): 2389–95. doi:10.1099/vir.0.80081-0. PMID 15269381.
- ^ Cox, Michael; Nelson, David R.; Lehninger, Albert L (2008). Lehninger principles of biochemistry. San Francisco: W.H. Freeman. ISBN 978-0-7167-7108-1.
- ^ Sagher, Daphna; Turkington, Edith; Acharya, Sonia; Strauss, Bernard (July 1994). «Production of UV-induced Frameshift Mutations in Vitro by DNA Polymerases Deficient in 3′ → 5′ Exonuclease Activity». Journal of Molecular Biology. 240 (3): 226–242. doi:10.1006/jmbi.1994.1437. PMID 8028006.
- ^ Johnson, Neil P.; Walter A. Baase; Peter H. von Hippel (March 2004). «Low-energy circular dichroism of 2-aminopurine dinucleotide as a probe of local conformation of DNA and RNA». Proc Natl Acad Sci U S A. 101 (10): 3426–31. Bibcode:2004PNAS..101.3426J. doi:10.1073/pnas.0400591101. PMC 373478. PMID 14993592.
- ^ Baase, Walter A.; Davis Jose; Benjamin C. Ponedel; Peter H. von Hippel; Neil P. Johnson (2009). «DNA models of trinucleotide frameshift deletions: the formation of loops and bulges at the primer–template junction». Nucleic Acids Research. 37 (5): 1682–9. doi:10.1093/nar/gkn1042. PMC 2655659. PMID 19155277.
- ^ a b Hu, J; Ng, PC (9 February 2012). «Predicting the effects of frameshifting indels». Genome Biology. 13 (2): R9. doi:10.1186/gb-2012-13-2-r9. PMC 3334572. PMID 22322200.
- ^ Tucker, Tracy; Marra, Marco; Friedman, Jan M. (2009). «Massively Parallel Sequencing: The Next Big Thing in Genetic Medicine». The American Journal of Human Genetics. 85 (2): 142–154. doi:10.1016/j.ajhg.2009.06.022. PMC 2725244. PMID 19679224.
- ^ Walsh, T.; Casadei, S.; Lee, M. K.; Pennil, C. C.; Nord, A. S.; Thornton, A. M.; Roeb, W.; Agnew, K. J.; Stray, S. M.; Wickramanayake, A.; Norquist, B.; Pennington, K. P.; Garcia, R. L.; King, M.-C.; Swisher, E. M. (2011). «From the Cover: Mutations in 12 genes for inherited ovarian, fallopian tube, and peritoneal carcinoma identified by massively parallel sequencing». Proc Natl Acad Sci U S A. 108 (44): 18032–7. Bibcode:2011PNAS..10818032W. doi:10.1073/pnas.1115052108. PMC 3207658. PMID 22006311.
- ^ Walsh, T.; Lee, M. K.; Casadei, S.; Thornton, A. M.; Stray, S. M.; Pennil, C.; Nord, A. S.; Mandell, J. B.; Swisher, E. M.; King, M.-C. (2010). «Detection of inherited mutations for breast and ovarian cancer using genomic capture and massively parallel sequencing». Proc Natl Acad Sci U S A. 107 (28): 12629–33. Bibcode:2010PNAS..10712629W. doi:10.1073/pnas.1007983107. PMC 2906584. PMID 20616022.
- ^ US Patent 5,958,684 (September 28, 1999) «Diagnosis of Neurodegenerative Disease» by Leeuwen et al
- ^ a b Harfe, BD; Jinks-Robertson, S (July 1999). «Removal of frameshift intermediates by mismatch repair proteins in Saccharomyces cerevisiae». Molecular and Cellular Biology. 19 (7): 4766–73. doi:10.1128/MCB.19.7.4766. PMC 84275. PMID 10373526.
- ^ Davies, J E; Rubinsztein, D C (2006). «Polyalanine and polyserine frameshift products in Huntington’s disease». Journal of Medical Genetics. 43 (11): 893–896. doi:10.1136/jmg.2006.044222. PMC 2563184. PMID 16801344.
- ^ Schmoldt, A; Benthe, HF; Haberland, G (1 September 1975). «Digitoxin metabolism by rat liver microsomes». Biochemical Pharmacology. 24 (17): 1639–41. doi:10.1016/0006-2952(75)90094-5. PMID 10.
- ^ Xu, XiaoLin; Zhu, KaiChang; Liu, Feng; Wang, Yue; Shen, JianGuo; Jin, Jizhong; Wang, Zhong; Chen, Lin; Li, Jiadong; Xu, Min (May 2013). «Identification of somatic mutations in human prostate cancer by RNA-Seq». Gene. 519 (2): 343–7. doi:10.1016/j.gene.2013.01.046. PMID 23434521.
- ^ «Cancer Genomics». National Cancer Institute at the National Institute of Health. Archived from the original on 18 March 2013. Retrieved 24 March 2013.
- ^ Ogura Y, Bonen DK, Inohara N, Nicolae DL, Chen FF, Ramos R, Britton H, Moran T, Karaliuskas R, Duerr RH, Achkar JP, Brant SR, Bayless TM, Kirschner BS, Hanauer SB, Nuñez G, Cho JH (May 31, 2001). «A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease» (PDF). Nature. 411 (6837): 603–6. Bibcode:2001Natur.411..603O. doi:10.1038/35079114. hdl:2027.42/62856. PMID 11385577. S2CID 205017657.
- ^ Farrell PM, Rosenstein BJ, White TB, Accurso FJ, Castellani C, Cutting GR, Durie PR, Legrys VA, Massie J, Parad RB, Rock MJ, Campbell PW (2008). «Guidelines for Diagnosis of Cystic Fibrosis in Newborns through Older Adults: Cystic Fibrosis Foundation Consensus Report». The Journal of Pediatrics. 153 (2): S4–S14. doi:10.1016/j.jpeds.2008.05.005. PMC 2810958. PMID 18639722.
- ^ Iannuzzi, MC; Stern, RC; Collins, FS; Hon, CT; Hidaka, N; Strong, T; Becker, L; Drumm, ML; White, MB; Gerrard, B (February 1991). «Two frameshift mutations in the cystic fibrosis gene». American Journal of Human Genetics. 48 (2): 227–31. PMC 1683026. PMID 1990834.
- ^ «Learning About Tay-Sachs Disease». National Human Genome Research Institute. Retrieved 24 March 2013.
- ^ Myerowitz, R (1997). «Tay-Sachs disease-causing mutations and neutral polymorphisms in the Hex A gene». Human Mutation. 9 (3): 195–208. doi:10.1002/(SICI)1098-1004(1997)9:3<195::AID-HUMU1>3.0.CO;2-7. PMID 9090523.
- ^ Truong, Hoa T; Dudding, Tracy; Blanchard, Christopher L.; Elsea, Sarah H (2010). «Frameshift mutation hotspot identified in Smith-Magenis syndrome: case report and review of literature». BMC Medical Genetics. 11 (1): 142. doi:10.1186/1471-2350-11-142. PMC 2964533. PMID 20932317.
- ^ Chung WK, Kitner C, Maron BJ (June 2011). «Novel frameshift mutation in Troponin C ( TNNC1) associated with hypertrophic cardiomyopathy and sudden death». Cardiol Young. 21 (3): 345–8. doi:10.1017/S1047951110001927. PMID 21262074. S2CID 46682245.
- ^ Hu, Hailiang; Gatti, Richard A (2008). «New approaches to treatment of primary immunodeficiencies: fixing mutations with chemicals». Current Opinion in Allergy and Clinical Immunology. 8 (6): 540–6. doi:10.1097/ACI.0b013e328314b63b. PMC 2686128. PMID 18978469.
- ^ European Patent [1] (December 10, 2003) «Use of coding microsatellite region frameshift mutation-derived peptides for treating cancer» by Bork et al
Further reading[edit]
- Farabaugh PJ (1996). «Programmed translational frameshifting». Annu. Rev. Genet. 30 (1): 507–28. doi:10.1146/annurev.genet.30.1.507. PMC 239420. PMID 8982463.
- Lewis, Ricki (2005). Human Genetics: Concepts and Applications (6th ed.). Boston MA: McGraw Hill. pp. 227–8. ISBN 978-0-07-111156-0.
- «Nylonase Enzymes». 20 April 2004. Retrieved 2 June 2009.
External links[edit]
- Frameshift+Mutation at the US National Library of Medicine Medical Subject Headings (MeSH)
- NCBI dbSNP database — «a central repository for both single base nucleotide substitutions and short deletion and insertion polymorphisms»
- Wise2 — aligns a protein against a DNA sequence allowing frameshifts and introns
- FastY — compare a DNA sequence to a protein sequence database, allowing gaps and frameshifts
- Path — tool that compares two frameshift proteins (back-translation principle)
- HGMD — Human Genome Mutation Database
2.8. Вариации генома человека: генные и хромосомные мутации
В основе практически всех генетических исследований лежит понятие вариации. Это понятие включает в себя все типы изменений последовательностей ДНК (мутаций), наблюдаемых на хромосомном или генном уровнях. С одной стороны, вариации генома служат объяснением межиндивидульного разнообразия, с другой, мутации могут приводить к патогенным изменениям жизнедеятельности организма, являясь, таким образом, причиной наследственного заболевания. Следует также ввести несколько терминов, использующихся для описания процесса мутационного изменения ДНК: локус – определенный участок хромосомы, содержащий специфические последовательности ДНК или гены, аллель – две или более альтернативных форм гена, расположенных в одном и том же локусе пары гомологичных хромосом. Если различие последовательности ДНК двух аллелей одного локуса наблюдается с частотой более 1 % в общей популяции, то данный тип вариации обозначается полиморфизмом. Изменение последовательности ДНК, имеющее меньшую частоту, как правило, называется мутацией. Известно два основных вида мутаций, связанных с наследственной патологией: хромосомные (геномные) – изменение числа и/или структуры хромосом (генома) в клетке и генные (изменение последовательности ДНК в конкретном гене). Исходя из данной классификации, можно выделить направления генетических исследований нарушений последовательности ДНК, приводящих к наследственным заболеваниям, которые изучает медицинская генетика, а именно, поиск изменений последовательностей нуклеиновых кислот и белков на молекулярном уровне (молекулярная генетика) и изучение изменений числа, структуры и организации хромосом (классическая и молекулярная цитогенетика).
Молекулярно-генетические исследования основаны на современных представлениях об особенностях молекулы ДНК и биохимических процессах транскрипции и трансляции. Основная их цель заключается в выявлении генных мутаций, приводящих к характерным фенотипическим проявлениям. Генные мутации представляют собой изменение расположения, потерю и приобретение ДНК относительно её линейной последовательности, обнаруживаемой в норме. Наиболее частые типы генных мутаций являются замена, потери и/или вставки одного нуклеотида. Последние обозначаются аббревиатурой SNP (single nucleotide polymorphsims) и проявляются наиболее часто в геноме человека. В среднем, SNP, ведущие к вариации между аллелями у одного индивидуума, встречаются в каждых 1500 пар нуклеотидов. Однако, большинство из них расположены в некодирущих последовательностях и, в основном, не имеют фенотипических последствий. Если изменение последовательности ДНК происходит в гене, кодирующем белок, то оно с высокой долей вероятности будет связано с нарушениями жизнедеятельности организма. Существует следующая классификация генных мутаций:
Миссенс мутации – замена одного нуклеотида на другой или несинонимические изменения последовательности ДНК. Теоретически можно выделить два типа подобных мутаций: консервативные и неконсервативные. Консервативные миссенс мутации приводят к замене одного кодона на равнозначный (кодоны, кодирующие один и тот же аминокислотный остаток) или на кодон другого аминокислотного остатка, который не изменяет физико-химические свойства белка, кодированного соответствующим геном. Неконсервативные миссенс мутации, как правило, изменяют биохимические свойства белка и, следовательно, приводят к нарушению его функциональной активности.
Нонсенс мутации – изменения кодирующей последовательности ДНК, приводящие к образованию стоп-кодона, вследствие чего синтезируется белок, в котором отсутствует какая-то часть его последовательности.
Мутация сдвига рамки считывания – любые изменения последовательности ДНК гена (в основном, потери или вставки нуклеотидов), которые приводят к сдвигу считывания последовательности в ходе транскрипции. Результатом этого является синтез совершенно нового белка или образование матричной РНК, не несущей в себе никакой информации относительно аминокислотной последовательности.
Непатогенные изменения последовательности ДНК – вариации последовательности ДНК, включающие консервативные миссенс мутации, или так называемые синонимические мутации, которые не изменяют закодированную информацию в ДНК гена или не воздействуют на функциональную активность белковых макромолекул.
Мутации также происходят в некодирующих последовательностях ДНК (интронах). Данный тип вариаций, как правило, не имеет фенотипических последствий. Тем не менее, при сдвиге рамки считывания или образовании альтернативных форм белковых макромолекул (альтернативный сплайсинг), эти вариации могут приводить к нарушению функциональной активности белковых макромолекул и, как следствие, фенотипическим последствиям. В данном контексте сложностью представляется идентификация патогенных мутаций, так как понятие «нормы» для медико-генетических исследований предположительно в силу того, что на молекулярном уровне геном человека является в значительной степени нестабильным. Иными словами, только рекуррентные мутации (наиболее частые повторные мутации, которые выявляются у индивидуумов с известным наследственным заболеванием) могут быть признаны патогенными. В случаях, когда обнаруживается новая мутация, возникает необходимость молекулярно-генетических исследований близких родственников пациента, чтобы определить является ли она причиной заболевания.
Хромосомные (геномные) мутации (аномалии) связаны либо с различными структурными перестройками хромосом, либо с изменением их числа (n). Численные изменения в наборе хромосом (кариотипе) могут быть двух типов: полиплоидии – умножение полного хромосомного набора (3n, 4n и т.д.) или генома, кратное гаплоидному числу хромосом; анеуплоидии – увеличение или уменьшение числа хромосом в наборе, некратное гаплоидному. Эти количественные изменения кариотипа обусловлены, как правило, нарушениями мейоза или митоза. Численные хромосомные аномалии в виде анеуплоидии делятся на моносомию (потерю хромосомы или её части – частичная моносомия) и трисомию или полисомию (приобретение одной/нескольких хромосом или её части – частичная трисомия). Данные изменения кариотипа связаны с комплексом врожденных пороков развития и, как правило, с заболеваниями, сопровождающимися умственной отсталостью, или тяжелыми психическими расстройствами. В настоящее время описаны случаи изменений хромосомного набора с участием половых хромосом и некоторых аутосом при шизофрении и аутизме. Например, до 5–15 % детей с аутистическими расстройствами имеют хромосомные аномалии. Это позволяет рассматривать хромосомный дисбаланс в организме в качестве одной из возможных причин отдельных случаев нервных и психических болезней.
Структурные изменения могут затрагивать всю хромосому, а также сопровождаться изменением количества генетического материала в ядре или его перемещением. Сбалансированные хромосомные аномалии представляют собой перестройки, за счет которых выявляется кариотип с измененным набором расположения генов в пределах хромосом или между хромосомами, который отличается от традиционного (нормального). В большинстве случаев носители сбалансированных хромосомных аномалий фенотипически нормальны, но для их потомства возникает большой риск иметь несбалансированный кариотип. Следует отметить, что в отдельных случаях носители сбалансированного кариотипа могут иметь различные врожденные пороки и/или микроаномалии, а также нарушения нервного и психического развития. Если при структурных хромосомных мутациях наблюдается потеря или приобретение генетического материала, то они являются несбалансированными хромосомными аномалиями.
Цитогенетически структурные хромосомные (геномные) перестройки классифицируют по принципу линейной последовательности расположения генов: делеции (потеря хромосомных участков), дупликации (удвоение хромосомных участков), инверсии (перевертывание на 180° относительно нормальной последовательности хромосомных участков), инсерции (вставки хромосомных участков) и транслокации (изменение расположения хромосомных участков). В последнее время в литературе хромосомные микроаномалии и перестройки могут обозначаться, как геномные.
Изменения генома (хромосом), приводящие к редким заболеваниям, могут включать как крупные микроскопически видимые перестройки (более 5 млн пн), так и вариации числа копий последовательностей ДНК (CNV) и однонуклеотидные полиморфные изменения последовательности ДНК (SNP). Как уже было сказано выше, в настоящее время для определения причины заболевания на геномном уровне используются различные технологии, наиболее распространенными из которых являются полногеномные методы, в частности, молекулярное кариотипирование (arrayCGH). Однако патогенность выявленных вариаций генома можно определить только при помощи использования инновационных биоинформатических технологий. Большой массив информации, собранный на интернет-ресурсах, позволяет уточнить функциональные особенности (онтологию) как отдельного гена, так и целой генной сети за счёт анализа последовательности кодируемого белка и моделирования молекулярных процессов, инициированных геномным изменением.
Таким образом, с хромосомными болезнями связаны аномалии микроскопически видимых численных или структурных нарушений хромосом, геномные же болезни связаны как с микроаномалиям хромосом, так и с вариациями числа копий последовательностей ДНК (CNV).
Большое значение имеет изучение хромосомных мутаций, под действием факторов внешней среды. Показано, что хромосомы человека отличаются высокой чувствительностью к действию радиации и химических веществ, которые принято называть мутагенными факторами (мутагенами). При анализе воздействия этих факторов следует различать нарушения в соматических и половых клетках. Первые затрагивают непосредственно жизнедеятельность исследуемого организма, тогда как вторые проявляются в последующих поколениях. Мутации хромосом в зародышевых клетках ведут к образованию аберрантных гамет, в результате которых возможна гибель зигот, эмбрионов на ранних стадиях внутриутробного развития, а также рождение детей со специфическими или неспецифическими хромосомными аномалиями, которые проявляются в виде определенной клинической картины или определенного фенотипа. Мутации хромосом в соматических клетках ведут к образованию неспецифичных хромосомных аномалий в виде хромосомных или хроматидных пробелов, разрывов, обменов в кариотипе, не ведущих к определенному фенотипу, характерному для конкретного заболевания. Подобные мутации не наследуются. Следует отметить, что при изучении такого рода воздействия мутагенных факторов представляется возможным оценить качественно и количественно действие ионизирующей радиации, химических веществ, вирусов, но полученные данные не могут быть перенесены на половые клетки, где результатом действия являются специфические хромосомные аномалии, влияющие на фенотип.
Хромосомные аномалии могут проявляться в так называемых мозаичных формах, к которым приводит неправильное деление клеток на различных стадиях эмбрионального и постнатального развития. Это позволяет разделить хромосомные аномалии на мозаичные и регулярные (аномальный кариотип наблюдается во всех клетках организма). Хромосомный мозаицизм представляет собой наличие нескольких популяций клеток с различным друг от друга хромосомным набором. Как правило, при мозаичных формах хромосомных аномалий наблюдают отсутствие отдельных клинических признаков определенного хромосомного синдрома и более легкое течение заболевания, но некоторые симптомы практически всегда присутствуют. Мозаичные структурные хромосомные аномалии наблюдаются достаточно редко, поэтому, когда речь идет о мозаичных хромосомных аномалиях, имеются в виду, в основном, численные аномалии, мозаичные формы которых имеют достаточно высокую популяционную частоту. Следует также отметить феномен тканеспецифического хромосомного мозаицизма – клетки с аномальным хромосомным набором присутствуют только в определенной ткани организма.
Чтобы разобраться, что такое генетическая мутация, вспомним, как устроены ДНК и гены.
ДНК (дезоксирибонуклеиновая кислота) — это длинная молекула, которую принято называть «двойной спиралью». Она хранит биологическую информацию, которая «записана» в виде генетического кода.
Ген — это основная «единица» наследственной информации. Он представляет собой кусочек ДНК.

Главный редактор, заведующий хирургическим отделением
Задать вопрос
Врач-хирург высшей квалификационной категории, доктор медицинских наук, профессор кафедры общей хирургии АГМУ.
Содержание
- Какова функция генов?
- Что такое мутация?
- Мутации: хорошие, плохие, нейтральные
- «Хорошие» мутации
- «Плохие» мутации
- «Нейтральные» мутации
- Что делать, если генетический тест показал мутацию?
- Мутации при раке
- Как часто в клетках тела человека происходят мутации?
- Почему мутации приводят к онкологическим заболеваниям?
- Протоонкогены
- Гены-супрессоры опухолевого роста
- Гены репарации ДНК
- Что способствует развитию мутаций, которые приводят к раку?
- Распространенные мутации при раке
- Почему важно изучать мутации при онкологических заболеваниях?
- Как определяют мутации при раке?
- Что такое эпигенетические изменения, и какую роль они играют в онкологии?
- Новости «Евроонко»
- Родословная нейронов: как носить в себе множество мутаций и выглядеть совершенно здоровым
- Автор
- Редактор
- Мутации: патология или норма?
- Сколько мутаций может содержать в себе геном нейрона?
- «Генеалогическое древо» нейронов
- «Движение — это смерть»
- Наследственные болезни человека
- Наследственные болезни человека. Классификация.
- Хромосомные болезни
- Генные болезни
- Заболевания с наследственной предрасположенностью или полигенные болезни
- Диагностика наследственных болезней
- Лечение наследственных болезней
- Читайте также:
- Как влияют соматические мутации на здоровье людей
- Что такое мозаицизм? Соматический мозаицизм и мозаицизм по половым клеткам
- Соматический мозаицизм
- Мозаицизм по половым клеткам
Какова функция генов?
В части генов в виде кода записаны «рецепты» изготовления белков. Именно белки выполняют основные функции для поддержания жизнедеятельности организма: они отвечают за пищеварение, кровообращение, иммунитет, передачу информации между клетками.
Код представляет собой последовательность нуклеотидов.
В нашей ДНК есть четыре азотистых основания:
Основания одной цепи соединяются с основаниями другой цепи парами (аденин с тимином, цитозин с гуанином).

Какие способности передаются по наследству?
Если посмотреть на двойную спираль ДНК, то ее горизонтальные «ступени» будут парами оснований, а вертикальные боковые части — сахарами и фосфатами.
Чтобы изготовить белки по записанному в генах коду, специальные соединения — ферменты — «читают» и копируют код. В результате получаются длинные одноцепочечные молекулы — РНК (рибонуклеиновые кислоты), но это еще не белок. РНК лишь несут в себе информацию о первичной структуре белка, поэтому их называют матричными (сокращенно — мРНК). Эти молекулы покидают ядро клетки и перемещаются в ее цитоплазму. Там специальные органы — рибосомы — считывают код мРНК и изготавливают по этому «рецепту» белок.
Что такое мутация?
Генетическая мутация — это любое изменение в нуклеотидной последовательности ДНК.
К основным типам мутаций относятся:
- транзиция — замена аденина на гуанин или замена тимина на цитозин;
- трансверсия — аденин или гуанин меняются местами с тимином или цитозином;
- делеция — потеря участка ДНК;
- инсерция — добавление участка ДНК;
- дупликация — удвоение участка ДНК;
- инверсия — изменение, при котором участок хромосомы поворачивается на 180°;
- транслокация — мутация, при которой хромосомы обмениваются фрагментами.
Мутации могут происходить по разным причинам.
Спонтанные генетические мутации
Они происходят на протяжении всей нашей жизни. Можно сказать, что это нормальное явление, которое случается в ходе разных процессов, например, при копировании ДНК.
Как правило, такие ошибки не грозят серьезными последствиями, потому что у нашего организма есть механизмы защиты.

Как связаны спортивные достижения и генетика
К ним относится, например, апоптоз — процесс программируемой гибели «испорченной» клетки, или репарация — починка нити ДНК. В этом случае ошибочный участок ДНК вырезается, а на его месте формируется новый.
Мутации, вызванные внешним влиянием
Мутации могут возникнуть под воздействием внешних неблагоприятных факторов, например, химических веществ, ионизирующего излучения или заражения вирусами.
Белки, которые отвечают за исправление ошибок, как правило, могут исправить испорченные цепи ДНК или привести одну хромосому в соответствие с другой. Но, если ошибки произошли на уровне генома или количества хромосом, защитные механизмы будут бессильны.
Наследственные генетические мутации
Такие мутации достаются человеку от родителей. Бывают случаи, когда генетическое нарушение передается из поколения в поколение (как, например, болезнь гемофилия), иногда мутации происходят в яйцеклетках и сперматозоидах и таким образом передаются ребенку.
Бывают случаи, когда мутации возникают на этапе формирования зиготы — клетки, которая образуется в результате оплодотворения. Как и в предыдущем случае, механизмы репарации с такими мутациями работают далеко не всегда, а ряд заболеваний и вовсе связан с нарушениями в процессе починки (например, пигментная ксеродерма — заболевание кожи, представляющее собой повышенную чувствительность к ультрафиолету).
Мутации: хорошие, плохие, нейтральные
Не все генетические мутации опасны. Важно понимать, что именно мутации объясняют генетические различия между видами. Изменения генов влекут за собой изменение характеристик организма, и в результате этого он может стать либо более, либо менее приспособленным к выживанию.
В ходе естественного отбора преимущество получают те живые существа, которые обладают более «полезным» набором характеристик, и тогда мутация закрепляется в популяции, становясь нормой.
«Хорошие» мутации
Ученым известно, что, например, у людей с определенным вариантом гена GPR75 риск ожирения снижен на 54%. А те, у кого есть хотя бы одна копия такого варианта гена, имеют более низкий индекс массы тела.

Мутации генов могут давать человеку и другие преимущества: так, мутировавший ген EPOR дал финскому лыжнику, трехкратному олимпийскому чемпиону Ээро Мянтюранта высокую чувствительность к эритропоэтину — гормону, который помогает нашим клеткам поддерживать оптимальный уровень кислорода и выводить углекислый газ. Это изменило и объем красных кровяных клеток в крови спортсмена, и объем кислорода, который эти клетки способны переносить. В результате Мянтюранта получил супервыносливость — его организм легко справлялся с повышенной потребностью в кислороде во время физических нагрузок.
«Плохие» мутации
Генетические мутации могут вызывать различные заболевания. Например, изменения гена DMD вызывают дистрофию Дюшенна — нервно-мышечное заболевание, которое проявляется у мужчин намного чаще, чем у женщин. А к серповидноклеточной анемии — нарушению в строении белка гемоглобина, который переносит кислород от легких к органам, — приводят мутации гена HBB. Хорея Гентингтона — тяжелое заболевание нервной системы — развивается из-за мутации в гене HTT.
Однако далеко не всегда генетическое заболевание связано с мутацией одного гена. Так, синдром Дауна возникает из-за изменения количества хромосом — в клетках пациентов с этой болезнью 47 хромосом вместо обычных 46.
Ряд заболеваний, таких как рак, диабет, расстройства аутического спектра, появляются из-за комбинации факторов. Пациенты могут иметь генетическую предрасположенность, но значительную роль играют и внешние факторы — неправильный образ жизни, неблагоприятная окружающая среда.
«Нейтральные» мутации
Нейтральные мутации, как следует из названия, не оказывают на здоровье человека ни положительного, ни отрицательного эффекта. Как правило, эти мутации затрагивают нуклеотидную последовательность ДНК, но не сказываются на строении белков.
Так происходит, потому что наш генетический код обладает так называемой избыточностью — это значит, что ряд аминокислот кодируется несколькими способами, чтобы случайные ошибки при копировании с меньшей вероятностью привели к нарушению функции или отсутствию кодируемого белка.
Бывает и так, что мутация гена все-таки меняет аминокислоту. Тем не менее, это не всегда приводит к нарушению функции белка.
Что делать, если генетический тест показал мутацию?
Во многих случаях наличие генетических нарушений не означает, что у человека непременно разовьется какое-то заболевание. Это значит лишь то, что пациент обладает генетическим вариантом, который чаще встречается у людей с этим заболеванием, и, вероятно, предрасположен к возникновению проблемы больше остальных.
Важнейшую роль в таких случаях играют внешние факторы: образ жизни, привычки, окружающая среда.
Ученые сходятся на том, что важными условиями сохранения здоровья являются:
● сбалансированное питание, богатое овощами и фруктами;
● регулярные занятия спортом;
● отказ от курения и алкоголя;
● достаточное количество сна.
Эти правила помогут значительно снизить риск развития таких распространенных заболеваний, как рак, проблемы с сердечно-сосудистой системой, диабет второго типа.

Генная терапия: шанс или фантастика?
В том случае, если у человека есть мутация, связанная с моногенным заболеванием (то есть таким, которое возникает из-за «поломки» всего лишь одного гена), то существует риск, что он передаст этот вариант гена своему ребенку. Кроме того, болезнь может проявиться и у самого обладателя «плохого» гена — в этом случае ему следует обратиться к специалистам. Как правило, генетические заболевания не лечатся, но врач сможет порекомендовать препараты или изменения образа жизни (например, диета), чтобы уменьшить проявления болезни.
Результаты Генетического теста Атлас подскажут персональные рекомендации по улучшению образа жизни, которые помогут минимизировать риск появления заболеваний. Используя эти знания, будет проще спланировать подходящий рацион, спортивные нагрузки и тренировки, профилактические обследования.
Мутации при раке
Тело человека состоит примерно из 37 триллионов клеток. Информация о строении и функциях каждой из них закодирована в ДНК. Любая злокачественная опухоль является результатом нарушения работы тех или иных генов, а главная причина этого кроется в мутациях. Некоторые из них человек получает с рождения, и они присутствуют во всех клетках тела. А некоторые возникают уже в течение жизни под влиянием тех или иных факторов — эти мутации будут обнаруживаться только в потомках той клетки, в которой изначально возникла «поломка».
На этой странице мы собрали всю информацию о генетических нарушениях, связанных с онкологическими заболеваниями, представленную на нашем сайте.

Как часто в клетках тела человека происходят мутации?
Мутагенез — процесс непрерывный. Он происходит на всех этапах развития любого организма: в половых клетках, с самых первых дней существования эмбриона и на протяжении всей жизни. К счастью, далеко не все мутации вредны. Многие из них нейтральные (то есть не приносят ни вреда, ни пользы), а некоторые даже дают организму определенные преимущества.
Мутации — это главный двигатель эволюции живых организмов. В 2018 году были опубликованы результаты исследования, во время которого ученые обнаружили, что у 20-летних людей на одну клетку слизистой оболочки пищевода в среднем приходится по 100 мутаций, а у людей более старшего возраста — по 2000. Большинство из них не опасны, но некоторые затрагивают онкогены.
Чаще всего рак связан именно с соматическими, приобретенными, мутациями. Согласно современным представлениям, наследственные мутации ответственны за развитие лишь 5–10% онкопатологий. А по результатам исследования, опубликованного в 2020 году, наследственные мутации, связанные с раком, встречаются у каждого восьмого онкологического больного.
Почему мутации приводят к онкологическим заболеваниям?
Конечно же, далеко не все мутации и далеко не во всех генах приводят к развитию онкологических заболеваний. Чтобы нормальная клетка стала злокачественной, нарушения должны произойти в определенных генах:
Протоонкогены
Это гены, которые в результате мутаций способны превращаться в онкогены. В свою очередь, онкогены — это дефектные гены, которые способствуют развитию злокачественной опухоли, например, путем бесконтрольного размножения клеток. Характерный пример — EGFR.
Гены-супрессоры опухолевого роста
В норме они «сдерживают» клетки и не дают им стать злокачественными. Когда в этих генах возникают мутации, они перестают выполнять свои функции. Например, к этой категории относится ген TP53, кодирующий белок p53.
Гены репарации ДНК
Чаще всего их относят к генам-супрессорам опухолевого роста, но иногда выделяют в отдельную группу. Белки, кодируемые этими генами, исправляют «ошибки», возникающие в ДНК. Например, продукты генов BRCA1 и BRCA2 восстанавливают двухцепочечные разрывы в ДНК путем гомологичной рекомбинации — процесса, при котором поврежденная хромосома использует свою «сестру-близнеца» в качестве шаблона для репарации. Когда эти гены перестают правильно работать из-за мутаций, ДНК не может нормально восстанавливаться, и в ней накапливается еще больше повреждений.

Что способствует развитию мутаций, которые приводят к раку?
Мутации, связанные с онкозаболеваниями, бывают двух основных видов. Наследственные мутации происходят в половых клетках, и затем они будут присутствовать во всех клетках тела ребенка. Соматические мутации присутствуют только в клетках, в которых они изначально возникли, и в их потомках — например, только в злокачественной опухоли.
Обычно, чтобы нормальная клетка превратилась в злокачественную, в ней должен возникнуть целый набор мутаций. В каждом конкретном случае невозможно точно сказать, что именно послужило причиной. Скорее всего, единой причины и нет. На организм человека постоянно действует множество факторов, и многие из них могут способствовать поломкам в генах.
Вот список некоторых распространенных факторов риска, способствующих развитию рака:
Некоторые инфекции, например, ВПЧ
Неблагоприятная экологическая ситуация, воздействие вредных веществ на работе
Пол — многие онкологические заболевания чаще встречаются у мужчин или женщин
Семейный анамнез: рак у близких родственников
Большое количество красного и обработанного мяса (говядина, свинина, баранина, фастфуд, сосиски и колбасы, бекон и пр.)
Распространенные мутации при раке
Мутации в гене EGFR — белка-рецептора эпидермального фактора роста, который находится на поверхности клеток и активирует их размножение
T790M — один из вариантов мутации в гене EGFR
Мутации в гене ROS1 — белка, который встроен в клеточную мембрану и передает сигналы, играющие роль в росте и дифференцировке клеток
Мутации в гене BRAF. Белок, который он кодирует, участвует в регуляции делений клеток путем активации специфического сигнального пути.
Слияние генов с участием NTRK — когда из двух генов получается “неправильный”, гибридный. Гены NTRK кодируют белки Trk, которые выполняют разные функции, в том числе защищают клетки от апоптоза.
Мутации в гене ALK — белка, встроенного в клеточную мембрану, который передает сигналы, связанные с ростом, миграцией клеток, образованием новых кровеносных сосудов
Мутации в генах BRCA — белков, которые помогают восстанавливать ДНК, когда в обеих ее цепочках происходят разрывы
Мутации в генах RAS — белков, которые передают сигналы внутри клеток и регулируют клеточные деления. Семейство RAS включает три гена: KRAS, NRAS и HRAS.
Мутации в PIK3CA — гене, который кодирует белок PI3K, участвующий в регуляции важных процессов в клетках
Мутации в HRR — группе генов, продукты которых участвуют в репарации ДНК при двухцепочечных разрывах
Мутации в TP53 — гене, кодирующем белок p53, «страж генома», который останавливает размножение клеток с поврежденной ДНК и «приказывает» им совершить «самоубийство».
Результатом некоторых мутаций может стать микросателлитная нестабильность — состояние, при котором нарушается восстановление ДНК, и она приобретает повышенную склонность к мутациям.
Почему важно изучать мутации при онкологических заболеваниях?
Для врачей-онкологов важно знать, какие мутации произошли в раковых клетках у конкретного пациента. Это помогает решать важные задачи:
- судить о степени агрессивности рака, выстраивать прогноз;
- определять тип, подтип некоторых злокачественных опухолей;
- подбирать наиболее эффективные противоопухолевые препараты;
- назначать персонализированную терапию при запущенном раке, когда не помогают стандартные схемы лечения из протоколов.
Выявление мутаций, связанных с раком, у здоровых людей помогает оценивать риск развития онкологического заболевания, проводить профилактику и решать, кому назначать дополнительные скрининговые исследования.
А ученым знания о мутациях в опухолевых клетках помогают создавать новые лекарства.


Как определяют мутации при раке?
В федеральной сети клиник экспертной онкологии «Евроонко» доступны все современные исследования для выявления мутаций при раке:
Что такое эпигенетические изменения, и какую роль они играют в онкологии?
Не меньшую (а может быть, даже и более важную) роль, чем мутации, в развитии рака играют эпигенетические изменения. Этим термином называют такие модификации, которые не меняют последовательность генетического кода, но влияют на активность генов.
Чаще всего встречаются две разновидности эпигенетических изменений (но есть и другие):
- Метилирование ДНК — присоединение к ее определенным участкам метильных групп. Чаще всего они заставляют «молчать» определенные гены. В норме у человека метилирован 1% всего генома. В некоторых раковых клетках этот показатель ниже. За счет этого в них могут «включаться» онкогены.
- Модификации гистонов. ДНК организована таким образом, что напоминает бусы — эта структура называется нуклеосомой. В качестве бусинок выступают особые белки — гистоны. Они обмотаны нитями ДНК и влияют на активность генов. Даже небольшие изменения в гистонах могут сильно повлиять на регуляцию работы генов, заставить некоторые из них «замолчать» или, напротив, активировать.

Эпигенетика — очень интересная наука. Возможно, со временем она поможет ученым создать еще больше эффективных препаратов для лечения рака.
Новости «Евроонко»

Почему у многих курильщиков не развивается рак легких? 20 апреля 2022

С возрастом у людей накапливается много мутаций, способных привес. 27 декабря 2021

Как родинка превращается в меланому? 08 декабря 2021
Лечение пациентов проводится в соответствии со стандартами и рекомендациями наиболее авторитетных онкологических сообществ. «Евроонко» является партнёром Фонда борьбы с раком. ВНИМАНИЮ ПАЦИЕНТОВ: Рекомендации по лечению даются только после консультации у специалиста. Ваши персональные данные обрабатываются на сайте в целях его корректного функционирования. Если вы не согласны с обработкой ваших персональных данных, просим вас покинуть сайт. Оставаясь на сайте, вы даёте согласие на обработку ваших персональных данных.
Политика конфиденциальности © ООО «Центр инновационных медицинских технологий». 2012 — 2022
Товарный знак зарегистрирован. Все права защищены. Незаконное использование преследуется по закону.




Содержание данного интернет ресурса (сайт https://www.euroonco.ru/), включая любую информацию и результаты интеллектуальной деятельности, защищены законодательством Российской Федерации и международными соглашениями. Любое использование, копирование, воспроизведение или распространение любой размещенной информации, материалов и (или) их частей не допускается без предварительного получения согласия правообладателя и влечет применение мер ответственности.
Сведения и материалы, размещенные на сайте , подготовлены исключительно в информационных целях и не являются медицинской консультацией или заключением. Авторы информационных материалов сайта не могут гарантировать применимость такой информации для целей третьих лиц и не несут ответственности за решения третьих лиц и связанные с ними возможные прямые или косвенные потери и/или ущерб, возникшие в результате использования информации или какой-либо ее части, содержащейся на сайте.
Сайт использует файлы cookies для правильного функционирования, индивидуального подбора контента в социальных сетях и сбора анонимной статистики о пользователях с помощью систем аналитики для повышения удобства использования. Оставаясь на сайте, вы соглашаетесь с правилами использования файлов cookies.
Родословная нейронов: как носить в себе множество мутаций и выглядеть совершенно здоровым

Автор
Редактор
Статья на конкурс «био/мол/текст»: На протяжении долгой истории развития нейробиологии ученые придерживались догмы: мозг взрослого человека не подвержен изменениям. Однако в ходе нового исследования впервые было показано, что значительное количество мутаций присутствует в мозговом веществе абсолютно здоровых людей, причем чаще всего они обнаруживаются в генах, которые нейрон использует наиболее активно. Попробуем разобраться, как этим можно воспользоваться и чем это грозит.
Обратите внимание!
Эта работа опубликована в номинации «лучшее новостное сообщение» конкурса «био/мол/текст»-2015.
Спонсором номинации «Лучшая статья о механизмах старения и долголетия» является фонд «Наука за продление жизни». Спонсором приза зрительских симпатий выступила фирма Helicon.
Спонсоры конкурса: Лаборатория биотехнологических исследований 3D Bioprinting Solutions и Студия научной графики, анимации и моделирования Visual Science.
Мутации: патология или норма?
Каждая клетка нашего тела была создана путем деления клеток-предшественниц, которые, в свою очередь, восходят в развитии к зиготе. Значит ли это, что общий путь развития всех клеток организма обеспечивает общность генетического материала? Нет, и виной тому — мутации (рис. 1).

Рисунок 1. «Древо развития» мутаций в организме человека. Нарушения, обнаруженные в коре головного мозга, часто встречаются и в периферических органах. Рисунок из [4].
Мутации — коварные преобразования ДНК, которые страшны тем, что могут возникать в клетках любых тканей многоклеточного организма и на любых стадиях его развития. Распространено мнение, что мутации опасны потому, что могут наследоваться потомством. Действительно, мутации, передающиеся по наследству, приводят к возникновению и развитию таких серьезных заболеваний нервной системы, как шизофрения, аутизм, болезнь Альцгеймера. Виной тому — приобретаемые детьми генетические нарушения половых клеток родителей. Однако существуют и другие, ненаследуемые мутации, которые возникают в соматических клетках человека на протяжении всей его жизни.
Большинство людей имеет определенное количество соматических мутаций. Известным примером следствий соматических мутаций является появление опухолевых клеток, для которых характерны генетические нарушения*. Однако далеко не всегда соматическая мутация приводит к развитию онкологических заболеваний. Часто изменения генома не выливаются в какие-либо серьезные заболевания и могут встречаться у полностью здоровых людей. До настоящего момента ученые точно не знали, накапливаются ли они в головном мозге в таком количестве, чтобы послужить причиной серьезных нарушений нервной системы.
По мере роста и взросления человека геномы нейронов его головного мозга накапливают существенные различия. К такому выводу пришли ученые Бостонской детской больницы (Boston Children’s Hospital) и Гарвардской медицинской школы (Harvard Medical School), опровергнув утверждение, что мозг взрослого человека не изменяется в течение жизни* [4, 5].
* — Последние годы оказались особенно урожайными на опровержения железобетонных нейробиологических догм. Как нам на радость разобрались с приговором «нервные клетки не восстанавливаются», описано в статье «Всё, что вы всегда хотели знать о взрослом нейрогенезе, но боялись спросить» [6]. — Ред.
Результаты недавнего исследования показали, что значительное количество соматических мутаций можно обнаружить в мозге полностью здоровых людей. Так, со временем геномы нейронов головного мозга человека начинают различаться — появляется мозаицизм. Это научное открытие позволит изучать роль соматических мутаций отдельных нейронов в развитии человека и ряда нервно-психических заболеваний.
Сколько мутаций может содержать в себе геном нейрона?
Ранее не было точно известно, способны ли соматические мутации, возникающие в нейронах головного мозга, провоцировать возникновение и развитие нейродегенеративных заболеваний. Для того чтобы установить истину, ученые решили изучить особую разновидность мутаций — однонуклеотидные варианты (single-nucleotide variants, SNVs). Эти нарушения могут возникнуть в нескольких или даже всего в одной клетке головного мозга. Исследователи проанализировали 36 нейронов, взятых из головного мозга трех умерших людей: 15-летней девушки, 17-летнего юноши и 42-летней женщины, которые не страдали нейродегенеративными заболеваниями.
Используя методы капиллярной цифровой полимеразной цепной реакции (digital PCR) и секвенирования геномов единичных клеток [7], ученые обнаружили, что каждый отдельный нейрон из трех образцов ткани мозга содержит в среднем от 1468 до 1580 однонуклеотидных вариантов (рис. 2). И если появление SNVs в опухолевых клетках связано преимущественно с ошибками при репликации ДНК, то нейронные мутации возникают в основном вследствие активной транскрипции генов.

Рисунок 2. Карта мутаций генома корковых нейронов одного человека. 136 нейронов головного мозга 17-летнего человека распределены по четырем группам (обозначены разными цветами), выделенным по одной или нескольким мутациям (буквами A-D обозначены 18 клональных соматических мутаций). Рисунок из [5].
Дополнительно ученые сравнили гены нервных клеток с генетическим материалом, взятым из других тканей — в частности, сердца и кожи. Этот анализ показал, что мутации в нейронах в целом совпадают с однонуклеотидными вариантами в других типах клеток, то есть такие мутации присутствуют и в нейронах, и в других частях организма человека. Более того, был установлен следующий интересный факт: в ряде случаев клетки коры мозга показывали более высокую степень родства не с соседними нейронами, а с другими клетками организма (например, кардиомиоцитами).
Также было проведено исследование нервных клеток, взятых из разных областей головного мозга, с целью обнаружения аналогичных мутаций. Полученные результаты позволили сделать предположение о происхождении нервных клеток.
«Генеалогическое древо» нейронов

Рисунок 3. «Родословное древо» человека из книги Э. Геккеля «Антропогения». Идея объединения всех живых существ в единое «древо» имеет более чем 150-летнюю историю. Рисунок с сайта vivovoco.astronet.ru.
Мутации возникают как за счет ошибок копирования ДНК, которые потенциально могут сопровождать каждый репликационный цикл, так и в результате иных мутационных процессов — например, под действием ультрафиолетового света. Закономерное следствие — каждая клетка организма может иметь свой собственный уникальный геном, который несет в себе информацию о происхождении и развитии клетки, воздействии на нее внешних факторов. Такие «записи» онтогенеза отдельных клеток позволят создать их «родословное древо».
В разных клетках происходят разные мутации, что обеспечивает несходство геномов. Кроме этого, мутационный профиль несет в себе долговременную память о происхождении и развитии каждой клетки. Информация, полученная при секвенировании геномов индивидуальных нейронов, может быть использована для декодирования всей картины развития человеческого мозга — для реконструкции своеобразного «генеалогического древа» нейронов. Этот подход позволит расширить знания о природе возрастных заболеваний и выявить различия между мозгом человека и мозгом других животных.
Основоположником генеалогии можно считать Чарльза Дарвина, который впервые изобразил филогенетическое древо живых организмов еще в 1837 году. В его основу легла идея о том, что все виды живых существ связаны друг с другом общим происхождением, подобно ветвям дерева, которые объединяет общий корень (рис. 3). Подобные мысли использовали при создании клеточной теории ученые Т. Шванн и М. Шлейден, определившие клетку как единый структурный элемент всех живых организмов. Наконец, более чем через 150 лет, в 2005 году, Д. Фрумкин и соавторы в своем исследовании показали, что соматические мутации присутствуют в клетках в достаточном количестве и могут быть использованы для воссоздания взаимосвязей всех клеток человека [8]. Таким образом, далеко не свежие идеи лежат в основе нового заключения о том, что каждый человек несет в себе собственное (клеточное) генеалогическое древо*.
* — Дерево — это красиво и понятно, дерево — это аллегория из мира эукариот. А как же работают биологи с прокариотическими дебрями, где схемы родственных связей не то что дерево не напоминают, даже лес для них простоват — сеть да и только? Об эволюционных перипетиях в разных мирах читайте: «Эволюция между молотом и наковальней, или как микробиология спасла эволюцию от поглощения молекулярной биологией» [9], «Карл Вёзе (1928–2012)» [10], «Вирусные геномы в системе эволюции» [11] и «Закинули археи эволюционный невод и вытянули. » [12]. — Ред.
Кристофер Уолш и другие сотрудники Гарвардской медицинской школы в результате исследования однонуклеотидных вариантов предложили подход к установлению происхождения нервных клеток человека [5]. Так, если в двух отдельно взятых нейронах присутствуют одни и те же мутации, то они с высокой долей вероятности происходят от одной клетки-предшественницы. В том случае, если совпадает лишь часть мутаций, пути развития нейронов в какой-то момент времени разошлись.
Сравнивая геномы нейронов и других клеток организма, можно сделать следующий вывод: если какая-то мутация присутствует и в головном мозге, и в других соматических клетках — она возникла на раннем этапе онтогенеза. Если же определенная мутация встречается лишь в некоторых нейронах, это говорит о том, что она появилась сравнительно недавно. Таким образом можно проследить «родословную» нейронов вплоть до конкретного дня эмбрионального развития.
«Движение — это смерть»
Выше упоминалось, что мутации, обеспечивающие различия геномов соматических клеток, могут быть вызваны многими факторами. Так, длительное время считалось, что основной причиной мутаций в клетках головного мозга являются ошибки репликации ДНК. Однако в результате настоящего исследования ученые установили, что нарушения возникают не во время деления клетки, а при экспрессии генов. Всем известный девиз «Движение — это жизнь» не работает в случае соматических мутаций нейронов. Исследователи установили, что каждый раз, когда гены нейронов нашего мозга начинают активно работать — запуская программу синтеза новых белков, — появляется определенный риск возникновения мутаций.
Ученые пришли к выводу о том, что мутации в головном мозге накапливаются с возрастом и могут быть причастны к развитию нейродегенеративных заболеваний. Получается, что любой человек, сколь бы здоровым он ни был, является носителем огромного количества соматических мутаций — своеобразных «факторов риска». Чем это реально может грозить и как этого избежать — покажет время и будущие исследования нейробиологов.
Наследственные болезни человека
Наследственные болезни человека это заболевания, связанные с нарушением работы наследственного аппарата клеток и передающиеся по наследству от родителей потомству. Основной резервуар генетической информации находится в ядерных хромосомах. Все клетки человеческого организма содержат в ядрах одинаковое количество хромосом. Исключение составляют половые клетки или гаметы — сперматозоиды и яйцеклетки, и малая часть клеток, которые делятся прямым делением. Меньшая доля генетической информации содержится в митохондриальной ДНК.
Наследственные болезни человека. Классификация.
Патология генетического аппарата бывает на хромосомном уровне, на уровне отдельного гена, а также бывает связана с дефектом или отсутствием нескольких генов. Наследственные болезни человека подразделяются на:
Хромосомные болезни
Наиболее известны хромосомные заболевания по типу трисомии — дополнительной третьей хромосомы в паре:
- Синдром Дауна — трисомия по 21 паре;
- Синдром Патау — трисомия по 13 паре;
- Синдром Эдвардса — трисомия по 18 паре хромосом.

Синдром Шерешевского — Тёрнера обусловлен отсутствием одной Х-хромосомы у женщин.
Синдром Кляйнфельтера — дополнительная Х-хромосома у мужчин.
Другие хромосомные болезни связаны со структурной перестройкой хромосом при их нормальном количестве. Например, потеря или удвоение части хромосомы, обмен участками хромосом из разных пар.

Патогенез хромосомных болезней не совсем ясен. По-видимому, срабатывает механизм «пятого колеса», когда отсутствие или лишняя хромосома в паре мешает нормальной работе генетического аппарата в клетках.
Генные болезни
Причины наследственных заболеваний на генном уровне заключаются в повреждении части ДНК, в результате которого возникает дефект одного определенного гена. Чаще всего генные мутации ответственны за наследственные дегенеративные заболевания или наследственные болезни обмена веществ в результате нарушения синтеза соответствующего структурного белка или белка-фермента:
- Муковисцидоз;
- Гемофилия;
- Фенилкетонурия;
- Альбинизм; ;
- Серповидноклеточная анемия;
- Непереносимость лактозы;
- Другие обменные заболевания.
Моногенные наследственные заболевания наследуются по классическим законам Грегора Менделя. Различают аутосомно-доминантный, аутосомно-рецессивный и сцепленный с полом типы наследования.

При близкородственных браках чаще всего реализуется именно генный тип наследственных заболеваний.
Заболевания с наследственной предрасположенностью или полигенные болезни
К ним относятся:
- ; ;
- Ишемическая болезнь сердца;
- Ревматоидный полиартрит;
- Рак молочной железы;
- Псориаз;
- Шизофрения;
- Аллергические заболевания;
- Язвенная болезнь желудка…
Список можно продолжать и дальше. Найдется лишь малая часть болезней, которые так или иначе не связаны с наследственной предрасположенностью. Действительно, все процессы функционирования организма обусловлены синтезом разнообразных белков, как строительных, так и белков-ферментов.

Но если при моногенных наследственных болезней за синтез соответствующего белка отвечает один ген, то при полигенных наследственных заболеваниях за сложный метаболический процесс отвечают несколько разных генов. Поэтому мутация одного из них может быть компенсированной и проявляться только при дополнительных внешних неблагоприятных условиях. Этим объясняется, что у больных данными заболеваниями дети болеют ими не всегда, и, наоборот, у здоровых родителей дети могут болеть этими болезнями. Поэтому в случае полигенных наследственных заболеваний можно говорить лишь о большей или меньшей предрасположенности.
Диагностика наследственных болезней
Методы диагностики наследственных болезней:
- . Большинство хромосомных и генных заболеваний диагностируются по внешним или клиническим признакам. Характерный внешний вид при синдроме Дауна, полидактилия при синдроме Патау, отсутствие пигментации при альбинизме, тяжелые формы дыхательной недостаточности при муковисцидозе.
- Генеалогический метод заключается в построении генеалогического древа на основании данных анамнеза. Позволяет рассчитать вероятность развития генных заболеваний у детей при болезни или носительстве мутировавших генов у родителей и предков.
- Лабораторная и инструментальная диагностика. Наследственные болезни человека, связанные на нарушением метаболизма, выявляются с помощью клинических анализов. Например, серповидноклеточная анемия по общему анализу крови, определением фенилаланина при фенилкетоурии, нарушение коагулограммы при гемофилии. При мраморной болезни выявляются характерные рентгенологические изменения костей, при гемофилии — гемартрозы.
- Цитогенетическое исследование идентифицирует количество и строение хромосом. Применяется для диагностики хромосомных болезней.
- Скрининг на наследственные заболевания ориентирован на выявление генетической патологии на доклиническом уровне. Это комплексный метод, заключающийся в проведении просеивающего теста на некоторые наследственные заболевания: муковисцидоз, фенилкетонурия, болезнь Тея-Сакса и некоторых других редких наследственных заболеваний.
- Пренатальная диагностика наследственных заболеваний — метод выявления наследственной патологии на стадии внутриутробного развития.
- Молекулярно-цитогенетические и молекулярно-биологические методы позволяют провести диагностику наследственных болезней на уровне дефекта гена. Перспективное направление, однако, оно значительно осложняется при полигенных наследственных заболеваниях, когда за проявление болезни отвечают множество разных генов. Даже при моногенных заболеваниях не всегда известен и идентифицирован ответственный ген, что также затрудняет диагностику.
- Методы генетического выявления предрасположенности и профилактика наследственных заболеваний в онкологии. В 2006 году в США была основана частная компания «23andMe». Главное направление деятельности компании — выявление степени предрасположенности к некоторым заболеваниям, в частности к раку молочной железы и яичников на основе анализа генов BRCA1 и BRCA2. В значительной мере интерес к этой теме был подогрет в 2013 году операцией по удалению груди известной голливудской актрисе А. Джоли.

Однако, следует учитывать, что мутации генов BRCA1 и BRCA2 ответственны за рак молочной железы (РМЖ) только в 5-10%, а их наличие или отсутствие лишь изменяет степень риска достаточно редкой формы РМЖ. Расчет эффективности этого метода будет представлен в следующих публикациях.
Лечение наследственных болезней
Симптоматическое лечение заключается в коррекции метаболических и других патологических нарушений, связанных с данным заболеванием.
Диетотерапия направлена на исключение продуктов, содержащих вещества, которые не усваиваются или не переносятся больными.
Генотерапия направлена на введение в генетический аппарат клеток человека, эмбриона или зиготы генетического материала, компенсирующего дефекты мутированных генов. Успехи генотерапии пока невелики. Но медицина с оптимизмом смотрит на развитие генноинженерных методов в терапии наследственных заболеваний.
Читайте также:
Антибиотики в продуктах
В мире от электрического тока ежегодно погибает 50 тысяч, а всего в ХХ веке от электричества погибло 2 или 3 миллиона человек. Интересно, придет ли кому-то в голову запр.
Отрицательный резус-фактор у женщин
Легкое конспирологическое чтиво о связи группы крови или резус-фактора с убывающим Сатурном и судьбой нисколько не отменяют проблему резус-конфликта и гемолитическую желт.
Дальтонизм или цветовая слепота
Ночью все кошки серы! Действительно, при недостатке освещения цветовые рецепторы сетчатки глаза не работают, и мы видим лишь оттенки серого. Но стоит взойти солнцу, и кра.
Как влияют соматические мутации на здоровье людей
Что такое мозаицизм? Соматический мозаицизм и мозаицизм по половым клеткам
Мозаицизм — присутствие в организме или ткани по крайней мере двух генетически отличающихся клеточных линий, производных от одной зиготы. Хотя мы имеем обыкновение считать, что при формировании клеток они получают одинаковый набор генов и хромосом, это упрощенное представление. Мы уже ввели понятие мозаицизма, вызванного инактивацией Х-хромосомы, формирующей две различных популяции соматических клеток у женщин, с активной отцовской или материнский Х-хромосомой.
Чаще мутации, возникающие в единственной клетке во внутриутробной или послеродовой жизни, могут вызывать линии клеток, генетически отличающихся от зиготы, поскольку однажды произошедшая мутация может передаваться всем потомкам клетки. Мозаицизм по числовым или структурным аномалиям хромосом — клинически важный феномен, а соматические мутации признают основными причинами многих типов опухолей.
Мозаицизм по мутациям в одном гене, в соматических или половых клетках, объясняет множество необычных клинических наблюдений, например сегментный нейрофиброматоз, когда кожные проявления появляются не по всему телу, а участками, или повторное рождение у здоровых родителей двух или более детей с несовершенным остеогенезом, высокопенетрантной аутосомно-доминантной болезнью.
Популяция клеток, несущих мутацию у мозаичного пациента, теоретически может присутствовать в некоторых тканях тела, но не в гаметах (чистый соматический мозаицизм), ограничиваться только гаметами (чистый половой мозаицизм) или присутствовать как в соматических, так и в половых клетках, в зависимости от того, когда произошла мутация в ходе эмбрионального развития. Включает ли мозаицизм только соматические ткани, только половые клетки или и те, и другие, зависит от времени появления мутации в эмбриогенезе — до или после разделения половых и соматических клеток.
Если до, то и соматические, и половые клетки будут мозаичными, а мутация может передаваться потомству и проявляться соматически в мозаичной форме. Мутацию, произошедшую позже, обнаруживают только в половых клетках или части соматических тканей. Таким образом, например, если мутация произошла в предшественнике половых клеток, часть гамет будет нести мутацию. До мейоза половые клетки проходят около 30 митотических делений у женщин и несколько сотен у мужчин, допуская массу возможностей для мутаций, происходящих в течение митотических этапов развития гаметы.
Выявление мозаицизма по мутации только в половых или соматических клетках может быть трудным, поскольку отсутствие мутации в клетках из легкодоступных соматических тканей (например, лейкоцитов периферической крови, кожи или клеток слизистой оболочки рта) не доказывает, что мутация не присутствует где-нибудь еще, включая половые клетки. Охарактеризовать распространенность соматического мозаицизма еще труднее, если мутантный аллель у мозаичного эмбриона встречается исключительно во внезародышевых тканях (т.е. в плаценте) и не присутствует в самом эмбрионе.

Соматический мозаицизм
Мутации, влияющие на морфогенез и проявляющиеся в ходе эмбрионального развития, могут быть обнаружены как сегментные или пятнистые аномалии, в зависимости от этапа, в котором произошла мутация, и происхождения соматической клетки. Например, нейрофиброматоз I типа иногда может проявляться как сегментный, влияя только на одну часть тела. Сегментный нейрофиброматоз I типа вызван мозаицизмом по мутации, произошедшей после зачатия. В таких случаях родители пациента здоровы, но если он (или она) рожает больного ребенка, фенотип у ребенка полный, т.е. не сегментный.
В таких случаях мутация находится в гаметах пациента и, по-видимому, произошла до разделения половой и соматической линии клеток.
Мозаицизм по половым клеткам
Так как шанс, что аутосомное или Х-сцепленное заболевание, вызванное новой мутацией, может неоднократно происходить в сибстве, очень низок, поскольку спонтанные мутации обычно происходят редко (порядка 1 на 104-106), появление двух независимых мутаций в том же гене в одной семье весьма маловероятно (менее чем 1 на 108-1012). После тщательного исключения даже малых проявлений болезни у здоровых родителей ребенка с аутосомно-доминантным или Х-сцепленным заболеванием и при отрицательных результатах молекулярного тестирования носительства обычно принято сообщать родителям, что болезнь их ребенка — результат новой мутации и шанс того же дефекта у последующего ребенка незначительный, равный популяционному риску.
Существуют, тем не менее, хорошо подтвержденные примеры, когда фенотипически здоровые родители с отрицательными тестами на носительство имеют более чем одного ребенка с высокопенетрантным аутосомно-доминантным или Х-сцепленным заболеванием. Такие необычные родословные могут объясняться половым мозаицизмом. Половой мозаицизм хорошо подтвержден почти в 6% летальных форм аутосомно-доминантного несовершенного остеогенеза, когда мутации в гене коллагена I типа приводят к формированию аномального коллагена, ломким костям и частым переломам.
Родословные, которые могут объясняться половым мозаицизмом, также отмечены при нескольких других заболеваниях, например гемофилии А, гемофилии В и мышечной дистрофии Дюшенна, но очень редко встречаются при других доминантных болезнях, например ахондроплазии. Точно измерить частоту полового мозаицизма сложно, но приблизительно считают, что самая высокая встречаемость отмечена при мышечной дистрофии Дюшенна, при которой до 15% матерей в изолированных случаях не имеют подтверждения мутации в их соматических тканях при наличии мутации в половых клетках.
Теперь, когда феномен полового мозаицизма признан, генетики и генетические консультанты отдают себе отчет о потенциальной погрешности прогноза, что специфический аутосомно-доминантный или Х-сцепленный фенотип, кажущийся новой мутацией, имеет незначительный риск повторения в потомстве. Очевидно, для болезней с доказанной возможностью полового мозаицизма фенотипически здоровым родителям ребенка, у которых предположительно болезнь возникла вследствие новой мутации, нужно сообщать, что риск повторения не настолько незначительный.
Кроме того, родители ребенка с любым аутосомно-доминантным или Х-сцепленным заболеванием имеют риск повторения 3-4%, даже если половой мозаицизм не доказан и если известно, что они не носители мутации. Таким парам следует предложить доступную пренатальную диагностику. Точный риск повторения оценить трудно, поскольку он зависит от доли мутантных гамет.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Мутации возникают в склонных к ошибкам процессах копирования, включающих РНК-посредника
Мутации возникают в склонных к ошибкам процессах копирования, включающих РНК-посредника
Мутацией называется изменение последовательности нуклеотидов в ДНК (рис. 2.6). Если мутация происходит в той области ДНК, которая кодирует белок, она изменяет триплетный кодон и может привести к замене аминокислоты, определяемой этим код оном. Таким образом, появление другой аминокислоты в белковой цепочке может быть вызвано изменением одного единственного основания в ДНК-последовательности (точковая мутация). Большая часть измененных белков функционирует ненормально (хотя иногда они и выполняют совершенно иную функцию, на ином физиологическом или метаболическом фоне). Поэтому с точки зрения дарвиновского «выживания наиболее приспособленного» большинство мутаций вредны и ставят клетку или многоклеточный организм в неблагоприятные условия при естественном отборе.
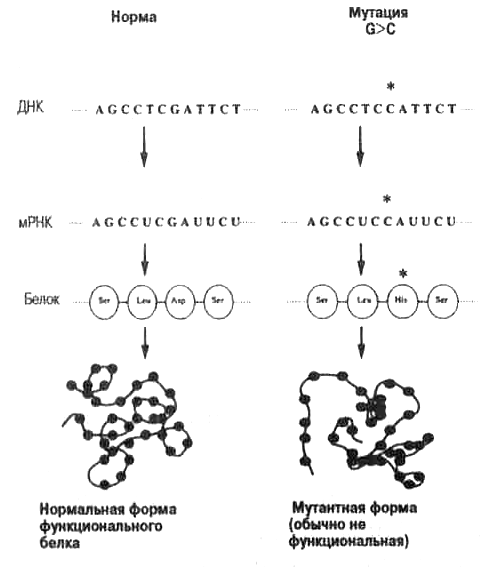
Рис. 5.1. Схема, показывающая как точковая мутация может привести к образованию нового мутантного белка с иной третичной структурой. Примечание: Дополнительную информацию см. в табл. 1.1, 5.1 и на рис. 2.5, 2.6 и 2.7.
Рис. 5.1 иллюстрирует эти рассуждения. На нем показаны нормальный и измененный белки. Обратите внимание, что одна единственная точковая мутация G -> С в кодоне, определяющем аспарагиновую кислоту (Asp), дает начало новому кодону, определяющему гистидин (His), в том же положении белковой цепочки (см. также приложение). Такая мутация может привести к радикальным последствиям для мутантного белка, который, складываясь, приобретает другую форму и, следовательно, — другую функцию (т. е. нормальная функция может быть утрачена). Этот принцип был продемонстрирован 40 лет назад, когда определили последовательность аминокислот бета-цепи гемоглобина здоровых людей и людей, страдающих серповидно-клеточной анемией. При этом наследственном заболевании нарушена способность гемоглобина переносить кислород. Молекула гемоглобина — гетеродимер, состоящий из двух альфа-цепей, или альфа-субъединиц, и двух бета-цепей, или бета-субъединиц. Заболевание проявляется у гомозиготных индивидов, у которых обе гомологичные хромосомы несут дефектный ген бета-цепи. Гетерозиготы, которые имеют одну нормальную и одну мутантную копию гена бета-цепи, болеют в легкой форме (так как половина их молекул гемоглобина способна нормально переносить кислород). Оказалось, что у больных серповидноклеточной анемией в 6-м положении бета-цепи вместо глутаминовой кислоты находится валин. И эта единственная замена в цепи из 146 аминокислот приводит к образованию больших агрегатов мутантных молекул гемоглобина в эритроцитах, деформирующих клетку так, что она принимает форму серпа. Интересно, что ген серповидноклеточной анемии сохраняется в Африке, потому что гетеро-зиготы имеют селективное преимущество при заражении малярией, так как плазмодий не может размножаться в серповидных эритроцитах столь же эффективно, как в нормальных.
Это опять возвращает нас к ключевому вопросу: как возникают генные мутации? Еще не так давно считалось, что они возникают «спонтанно» под влиянием космических лучей, рентгеновского излучения или ультрафиолетового света. Чарлз Дарвин называл такого рода изменения «спортами» и предполагал, что их случайно порождают условия Природы. Случай действительно играет роль, — разные мутации появляются с разной частотой. Однако сейчас, после 30—40-летнего периода накопления данных по вирусологии и молекулярной биологии, тайн вокруг причин появления мутаций гораздо меньше. В начале 1980-х гг. Дарил Ренни (Renney) из Университета Ла Троуб (Мельбурн) провел очень полезный анализ этой проблемы [7]. Благодаря его работе и выполненным ранее исследованиям Нобелевских лауреатов Артура Корнберга (Komberg), Манфреда Айгена и Говарда Темина (открывшего обратную транскрипцию у РНК-содержащих опухолевых вирусов) и вирусолога Джона Холленда (Holland), мы имеем логически последовательный способ анализа механизмов возникновения генных мутаций. Все дело в точности копирования ДНК- или РНК-последовательностей по матричным молекулам ДНК или РНК, которые осуществляются четырьмя ферментами, копирующими нуклеиновые кислоты: ДНК-полимеразой, РНК-полимера-зой, РНК-репликазой и обратной транскриптазой.
Исследования на молекулярном уровне показали, что ферменты, участвующие в репликации ДНК, способны к редактированию и исправлению ошибок. Возникновение мутаций в ходе репликации ДНК — редкое событие (рис. 5.2). Максимальная частота таких мутаций, вероятно, меньше, чем 10-8, а истинная частота ошибок, вероятно, еще меньше — около 10-10 (меньше, чем одна на 10 миллиардов реплицированных оснований). Чрезвычайно высокая точность копирования информации обеспечивается ДНК-полимеразой («машиной, копирующей ДНК»), которая по мере продвижения вдоль матричной ДНК-цепи проверяет, нет ли ошибок во вновь синтезированной копии. О наличии ошибок она «узнает» по искажению двойной спирали ДНК, которое имеет место, если Т соединится с G или С с А. Обнаружив такой участок, ДНК-полимеразный ферментный комплекс вырезает неправильное основание (или группу оснований) и вставляет то, которое должно быть на этом месте (законное основание). Скорость точной репликации у бактерий примерно 500 оснований в секунду, а у высших клеток (включая клетки человека) около 50 оснований в секунду. ДНК хромосом высших клеток много длиннее, а сами хромосомы устроены намного сложнее, чем маленькие и простые бактериальные геномы. У высших клеток, в отличие от бактерий, ДНК в хромосомах образует комплекс с белками (гистонами), которые участвуют в сворачивании длинных нитей ДНК в серию петель, для того чтобы их можно было упаковать внутри ядра. Репликация ДНК начинается одновременно в нескольких сайтах (точках) каждой хромосомы, поэтому большой набор ДНК-последовательностей реплицируется за 5—20 ч.
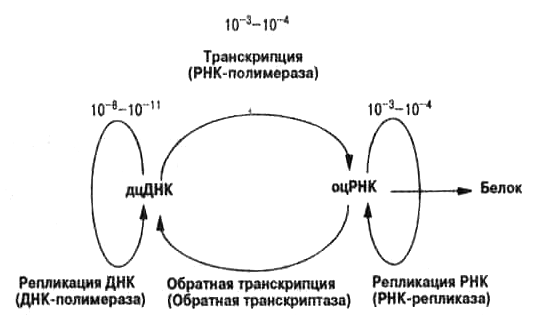
Рис. 5.2. Частота ошибок при синтезе ДНК и РНК. Примечание: о частоте ошибок судят по частоте включения неправильного основания на одно основание за одно событие копирования (см. также рис. 2.4); дц = двухцепочечная, оц = одноцепочечная.
Вспомним, что в гл. 2 мы уже обсуждали высокий уровень ошибок при образовании РНК по матрице ДНК (транскрипции) и при образовании ДНК по матрице РНК (обратной транскрипции). Оба этих типа копирования характеризуются частотой точковых мутаций Ю-3—Ю-4, что существенно выше, чем частота ошибок при репликации ДНК (от Ю-8 до Ю-9). Неточность, большое число ошибок имеют место и при репликации генома РНК-содержащих вирусов, например, вируса гриппа. Этим объясняется быстрое генетическое изменение вируса, приводящее к пандемиям гриппа. В жизненном цикле вируса СПИДа (ВИЧ) чередуются неточные процессы копирования РНК -> ДНК (на стадии интеграции) и ДНК -> РНК (на стадии экспрессии в течение инфекционного цикла). Для этого вируса также характерна высокая частота мутаций. Таким образом, все процессы копирования, включающие одноцепочечные РНК-посредники (превращение РНК в ДНК и наоборот), идут с большим числом ошибок, при этом репарация последовательности невозможна, поскольку ферменты, осуществляющие такое неточное копирование полинуклеотидов (РНК-полимераза, обратная транс -криптаза и РНК-репликаза), как оказалось, не имеют функций проверки и исправления ошибок.
Все сказанное выше означает, что какая-то доля мутаций в ДНК может возникать в результате ошибок копирования, включающего промежуточные РНК-посредники (рис. 5.2).
Мутации, которые передаются потомкам, появляются с низкой частотой. Они возникают в половых клетках и называются генеративными. Это редкие ошибки, которым удалось ускользнуть от «проверок» ДНК-полимеразы во время репликации ДНК и упаковки ее в гаметы самцов и самок (сперматозоиды и яйцеклетки). Они могут вызывать дефекты (т. е. влиять на фенотип и состояние здоровья индивида). Например, в случае серповидцоклеточной анемии такая мутация обуславливает тяжелую патологию у гомозигот (у которых обе копии гена дефектны) и более легкую форму болезни у гетерозигот, потому что белковый продукт нормальной копии гена частично компенсирует отрицательный эффект дефектной копии.
Однако существуют варианты некоторых генов (альтернативные формы генов называют аллелями), которые не влияют на здоровье индивида, у которого они проявляются. Эти варианты составляют нормальную изменчивость в популяциях организмов, существующую, по предположению Дарвина, до того, как начинает действовать естественный отбор. Важный вопрос: как появляются эти «добрые» аллели?. Согласно неодарвинистским представлениям, все эти аллели возникли в результате случайных мутаций в ДНК зародышевой линии и сохранились в популяции (так называемом «пуле генов») вследствие естественного отбора. В гл. 7 мы постараемся дать альтернативное объяснение этого феномена в рамках теории обратной связи соматических и половых клеток.
Предполагают, что рост числа врожденных аномалий и спонтанных абортов вызван факторами окружающей среды, такими как загрязнение токсическими химическими веществами. Например, резкое повышение частоты врожденных аномалий зарегистрировано в городах, расположенных вокруг сильно загрязненного, гибнущего Аральского моря. Такие же данные имеются относительно ветеранов вьетнамской войны и жителей северного Вьетнама, подвергшихся воздействию токсичных дефолиантов. Вещества, которые действуют на гены, изменяя кодирующую ДНК-последовательность, называются мутаге-нами. Возможно, их действие основано на том, что они нарушают нормальный процесс репарации. Установлено, что в клетках бактерий и эукариот, в которых индуцировано большое число повреждений ДНК, включается склонная к ошибкам репарация.
Читайте также
Глава V Как возникают различия между клетками
Глава V
Как возникают различия между клетками
Проблема возникновения различий между клетками имеет длительную историю: попытки ее экспериментального решения предпринимались еще в конце прошлого столетия, а теоретические споры преформистов и эпигенетиков восходят к
1. Роль гормонов в процессах развития
1. Роль гормонов в процессах развития
Если гормон равномерно распределяется по всему организму, то он принципиально не может создавать различий между совершенно одинаковыми клетками. Действительно, если клетки одинаковы, то и их реакция на любые внешние воздействия, в
Спонтанные мутации
Спонтанные мутации
О внезапных изменениях наследственности знал еще Дарвин. Взгляните на рисунок, где изображены овцы. Слева овца нормальная, в центре и справа овца и баран с сильно укороченными ногами. Сперва появилось одно-единственное такое животное. Изменение
Глава 5 СОМАТИЧЕСКИЕ МУТАЦИИ
Глава 5
СОМАТИЧЕСКИЕ МУТАЦИИ
В гл. 4 мы высказали предположение, что соматические мутации V-генов играли важную роль в эволюции иммунной системы позвоночных. Сформулируем теперь несколько вопросов и постараемся ответить на них. Вопрос первый: каким образом создается
Микробы и мутации
Микробы и мутации
Рассказ о трансформации, трансдукции и конъюгации должен был убедить читателя, что микроорганизмы действительно могут изменять свою природу. Во всех рассмотренных случаях микробиологи использовали влияние генетического материала (ДНК) одних микробов
Раковые клетки возникают из клеток собственного тела. Значит, они свои, а не чужие. Значит, иммунная система не может их «увидеть».
Раковые клетки возникают из клеток собственного тела. Значит, они свои, а не чужие. Значит, иммунная система не может их «увидеть».
— Иммунная система организма направлена на то, чтобы уничтожать любые клетки, которые были или стали чужеродными, не так ли?
Рак,
2.1. Наследственность и ее роль в процессах роста и развития
2.1. Наследственность и ее роль в процессах роста и развития
Наследственностью называется передача родительских признаков детям. Некоторые наследственные качества (форма носа, цвет волос, глаз, очертания лица, музыкальный слух, певческий голос и др.) не требуют для своей
Хромосомные аномалии и мутации
Хромосомные аномалии и мутации
Факты, которые мы приводили выше, известны ученым уже довольно давно. Сопоставлением и изучением этих фактов и занимается генетика — наука о явлениях наследственности и изменчивости. Основное положение менделевской генетики — учение о
Полезные мутации переключателей
Полезные мутации переключателей
Устойчивость к ядам, вирусам, бактериям и прочим паразитам, защитная окраска, превращение опадающих семян в неопадающие — все это примеры простых адаптаций, для развития которых бывает достаточно одной-двух удачных мутаций, поддержанных
5.1. Мутации
5.1. Мутации
Теория мутаций составляет одну из основ генетики. Ее основные положения были разработаны голландским ученым Г. де Фризом еще в начале XX в.Мутации – это наследственные изменения генетического материала. Они характеризуются как редкие, случайные,
Мутации
Мутации
Мутация — внезапное изменение гена. Она проявляется в первом же поколении потомков, если мутантный ген будет доминантным. Но рецессивный ген — мутант может скрытно наследоваться в течение нескольких поколений до тех пор, пока в родительскую пару не подберутся
Мутации с материнским эффектом
Мутации с материнским эффектом
У таких разных организмов, как морские ежи и лягушки, события, происходящие на ранних стадиях дробления, и, в сущности, большая часть, если не все развитие, предшествующее гаструляции, не зависят от генома зиготы. Информацию, необходимую для
Мутации, затрагивающие органогенез
Мутации, затрагивающие органогенез
Как мы убедились выше, события, происходящие на ранних стадиях развития, в значительной степени зависят от информации, поставляемой материнским организмом. Однако примерно ко времени гаструляции важную роль в дальнейшем развитии
Гомеозис и гомеозисные мутации
Гомеозис и гомеозисные мутации
Действие генов теснейшим образом связано с онтогенезом, и эта их связь выявляется при возникновении мутаций, которые резко прерывают развитие организма. Существуют, однако, мутации другого класса, которые изменяют процесс онтогенеза, но
8.2. Мутации
8.2. Мутации
Если бы организмы развивались поколение за поколением в одних и тех же окружающих условиях и передавали своему потомству все время одни и те же гены, сочетание эффектов генетической наследственности и морфического резонанса привело бы к бесконечному
Враг №2: активные формы кислорода Самый распространенный мутаген. Хитрость заключается в том, что эти активные формы генерируются в ходе самых обычных химических реакций, которые протекают в человеческом теле. Тут может возникнуть недоумение, ведь кислород — это газ, который содержится в атмосфере нашей планеты, с ним ассоциируется дыхание полной грудью, свежесть и еще какие-то приятные ощущения из рекламы стиральных порошков.
Разгадка кроется в названии. Кислород окисляет вещества, которые встречает на своем пути. Вспомните перекись водорода, которую выливают на разбитую коленку — примерно то же самое происходит в клеточных масштабах при выделении активных форм кислорода. Активные формы кислорода разрушают мембраны, из которых построены живые клетки, выдергивают отдельные основания из цепи ДНК и вносят разрывы. Страшно не только то, что они делают, но и то, как изощренно это происходит. Поскольку выделение АФК осуществляется постоянно, при поломке нейтрализующего их механизма клетки постоянно подвергаются бомбардировке мутагенами и гибнут в муках.
Доказано: от окисления и активных радикалов спасают антиоксиданты. Это вещества, которые содержатся в свежих ягодах и фруктах, зеленом чае, орехах и красном вине. Они переводят радикалы в неактивную форму. Иными словами, антиоксиданты — это такие альтруисты. Они выходят на улицу, видят, как хулиганы разрушают стены, и принимают удар на себя. Антиоксиданты обладают множеством чудесных свойств, одно из которых — защита от старения. Согласно некоторым гипотезам, старение ассоциировано с выделением АФК. Отдельно выделяется легион — витамин Е — собирательное название для нескольких жирорастворимых веществ, обладающих антиоксидантной активностью. При поступлении в пищеварительную систему ингредиенты витамина Е проходят отбор в печени, и уже в круг обмена веществ вступает в основном альфа-токоферол.
Важно учитывать собственную генетическую предрасположенность к усвоению витаминов, так как антиоксиданты при чрезмерном потреблении вредны: они могут препятствовать усвоению других микроэлементов.
Враг № 3: афлатоксины. Название принадлежит группе ядов-канцерогенов, вырабатываемых некоторыми видами плесени. Афлатоксины — это ответ на детский вопрос: «Почему нельзя есть землю и опавшие листья?», на взрослый вопрос: «Почему у чая есть срок годности?» и на старческий вопрос: «Почему нельзя есть заплесневевшую крупу?» Когда условия хранения не соблюдаются, на продукте, как на питательной среде, растут плесневые грибы рода Aspergillus. Опасность может подстерегать в крупах, специях, орехах, чае, молоке, яйцах, мясе, сухофруктах, хлебе и промышленных соках. Плесень может вырасти в результате неправильного или длительного хранения, а ароматизирующая добавка заглушит неприятный запах. Если доза афлатоксинов не смертельная, но регулярная, это может послужить причиной для развития цирроза и рака печени. Особую опасность продукты с афлатоксинами представляют для будущих матерей: помимо общего отравления, афлатоксины могут нарушить эмбриогенез.
Доказано: полезные пищевые привычки помогут избежать этого врага. Будьте аккуратны и не ешьте просроченные продукты. Старайтесь тщательно мыть продукты и готовить еду непосредственно перед употреблением. Впрочем, люди с непереносимостью глютена, лактозы или кофеина в отношении афлатоксина чувствуют себя немного спокойнее: больше половины продуктов из группы риска уже исключены из их меню. Такая особенность пищевого поведения, как привычка переедать, может быть вдвойне вредна в данной ситуации.
Враг №4: бензол. Бензол — химическое соединение, без которого невозможно представить современную жизнь. Это токсин и канцероген, который входит в состав нефти и бензина, а также широко применяется в производстве лекарств, пластмасс, резины и красителей. Если поместить человека в замкнутое пространство и постепенно закачивать туда бензол, то сначала он почувствует эйфорию. Затем появятся сонливость, тошнота, головная боль, мышечные подергивания. Если оставить испытуемого в этой камере на длительное время, то он умрет, если же его выпустить, то можно будет наблюдать целый комплекс расстройств, вызванных отравлением бензолом. Однако нам интересно не это. Бензол часто встречается в некачественной косметической продукции и пластмассовых изделиях, то есть не исключено систематическое отравление бензолом в небольших количествах, что может провоцировать возникновение различных видов гемато-онкологических заболеваний.
Доказано: витамины группы В — Ниацин, фолат и кобаламин — необходимы для репарации ДНК. Дефицит этих витаминов в первую очередь заметен при отравлении бензолом. Зачастую люди получают витамины этой группы с мясом животных. Подробнее узнать о том, на какие процессы влияют витамины группы В и как диагностировать их недостаток, можно по ссылке.
Берегите себя. Враги в лице мутагенов могут быть повсюду. Когда речь заходит о здоровом образе жизни, тем, кто пытается изменить все своими силами, приходится балансировать между массой советов и здравым смыслом. Граница зыбкая и устоять на ней больше шансов у того,, кто знает себя и не наносит себе вред.